
光催化还原CO2计算模拟培训班
随着环保意识的增强和对新型能源需求的日益增加,光催化还原二氧化碳技术的研究受到广泛关注。在这一领域,第一性原理计算软件VASP能够模拟和分析材料的结构、能带、电子密度和热力学性质等,为理解材料本质提供有效的途径!VASP还能帮助研究者探索材料吸附CO2分子的机制、分析光吸收等性质,并优化电子结构,提高催化性能。因此,VASP计算在光催化还原二氧化碳研究中的重要性不断增加,为开发高效、环保的新型能源提供理论依据。
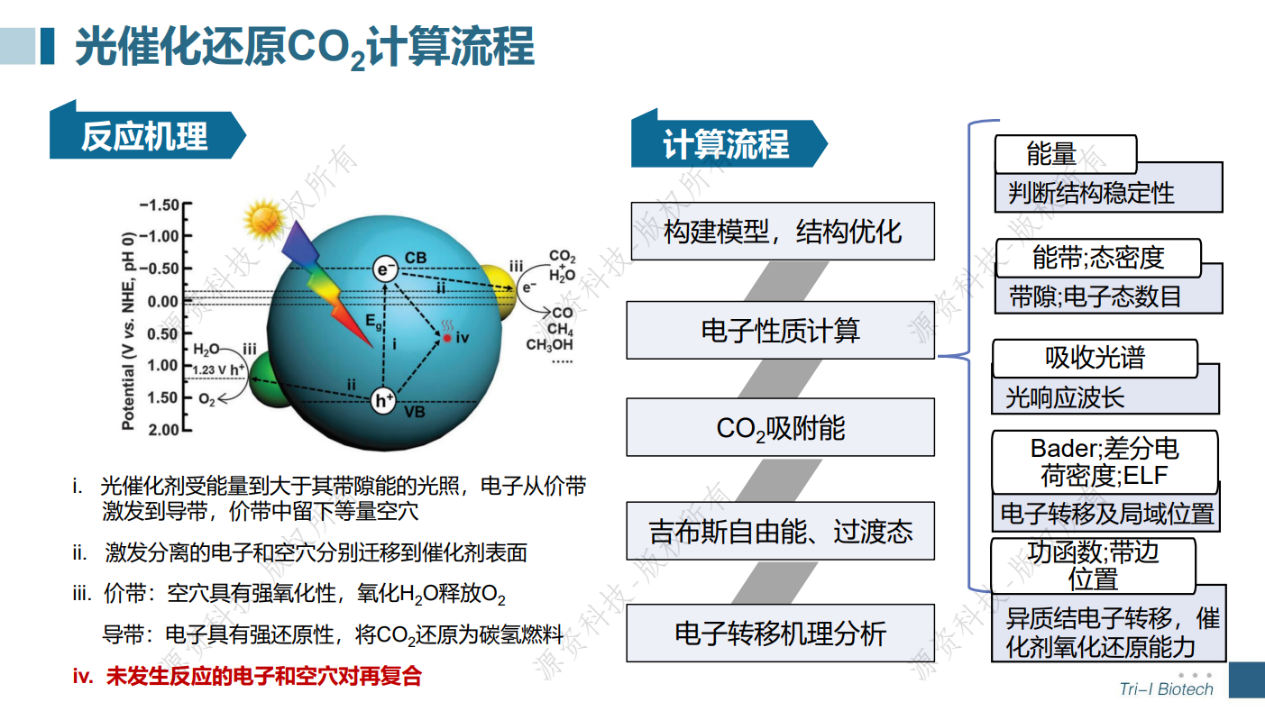
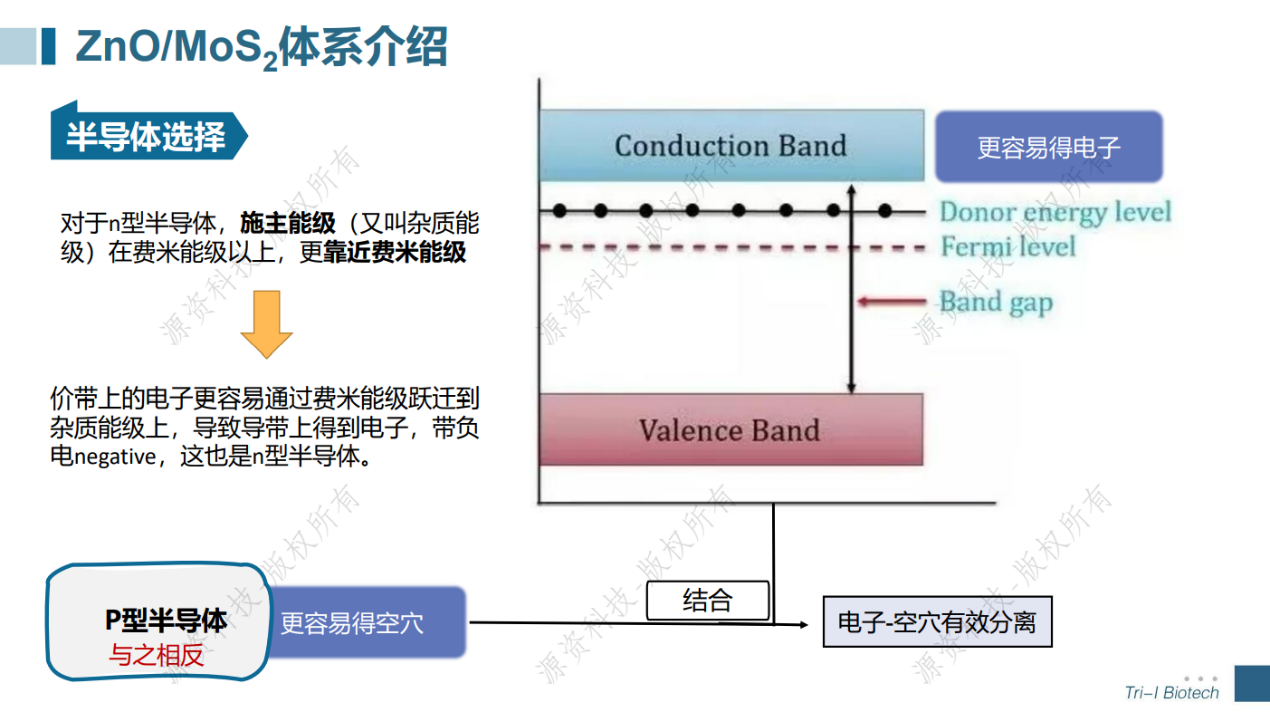
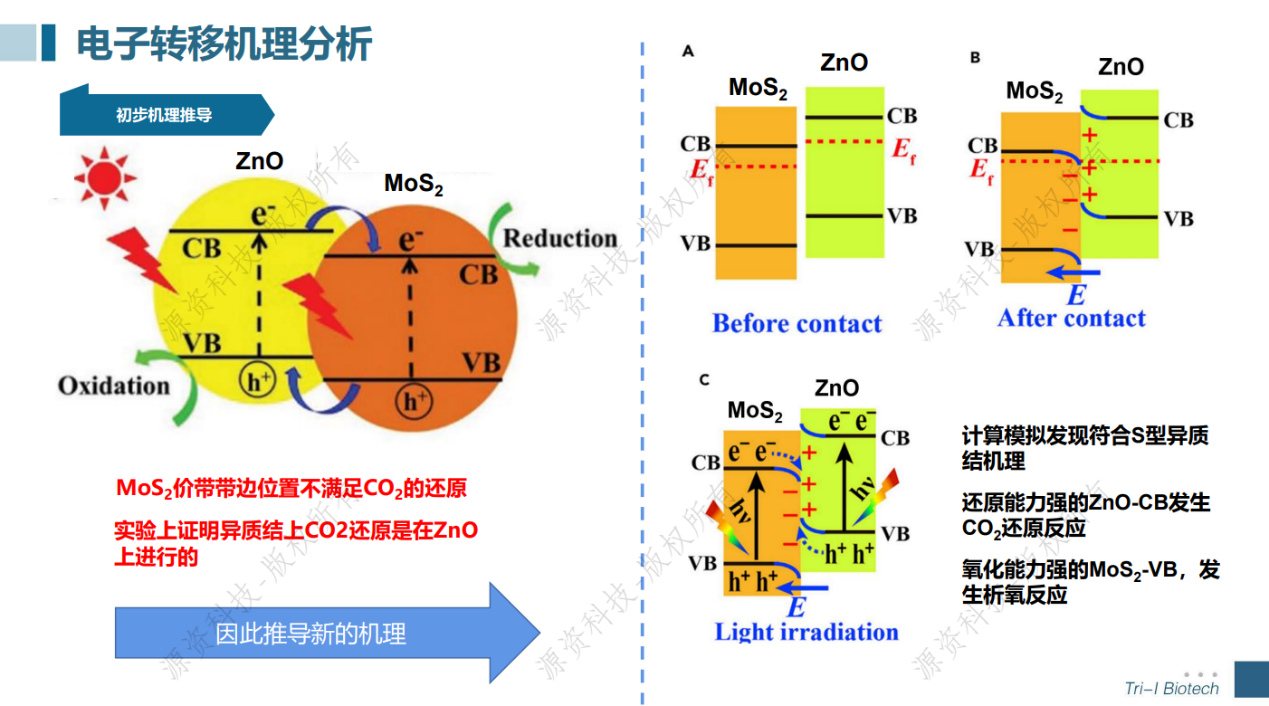
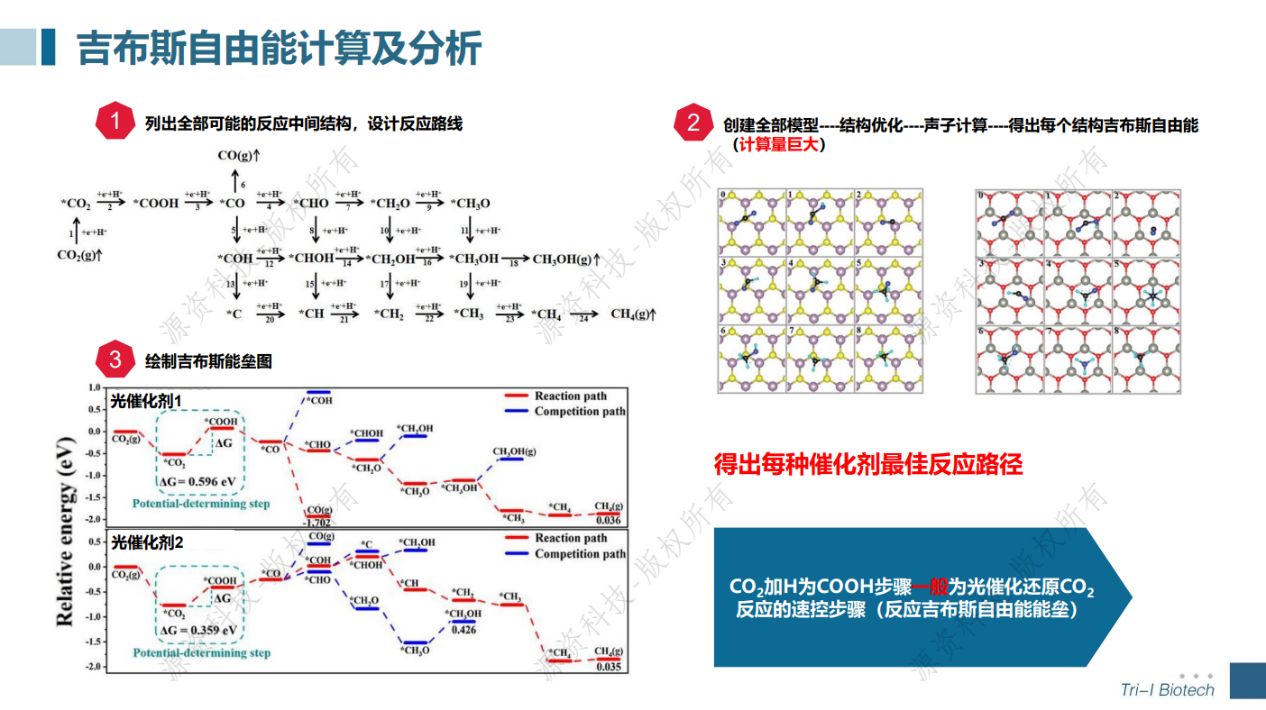
光催化还原CO2计算模拟专题速成班已于7月18日顺利举办。在短短一天的培训当中学员不仅学会了晶胞、表面、超胞、CO2吸附结构以及异质结结构的建模,还跟着讲师一起计算得到了光催化剂的能带、态密度、电荷密度、带边位置、反应吉布斯自由能等。讲师与学员积极互动交流,获得了一致好评!
精彩内容分享
Q&A答疑分享
1. VASP计算时哪些情况需要计算自旋极化?哪些情况需要加U?
A:需要计算自旋极化的情况包括: 系统含有磁性原子或磁性离子;系统处于磁场中;系统具有自旋序(例如磁性体中的铁磁序、反铁磁序等)。
需要加U的情况包括:对于在一些过渡金属氧化物(TM oxides)的计算中,需要加入U参数;一些具有强关联的体系,如强关联系统(如镍三氧化物等);被认为具有d或f电子的原子,如铁、钴、镍、钒、铬等过渡金属。
需要注意的是,U的具体取值需要参考对应体系的文献或者通过计算测试来确定,不能一概而论。同时,U的取值也影响着计算结果的可靠性和准确性。
2. 什么情况下需要加赝氢?
A:表面结构如果为极性表面,在优化过程中,为了防止极性面上下层电荷之间的物理性转移,其内部残余电场需通过赝氢饱的方式进行淬灭。
3. MedeA-vasp计算二维材料的光学性质,如何使用BSE方法修正?
A:MedeA-VASP使用BSE方法修正光学性质计算,需要将计算参数ALGO = BSE参数加入到Add to input面板当中以启用BSE计算。
4. VASP计算时Reciprocal space和Real space对计算结果影响大吗?一般计算体系多少个原子以上的时候要选用Real space?
A:VASP计算时选择使用Reciprocal space(k点网格)或Real space(实空间网格)取决于所要计算的体系的特定情况,以及需要得到哪些信息。
在Reciprocal space中,计算结果通常更加准确,尤其是对于周期性材料,通过合理选取k点数,可以得到更准确的计算结果。在一些情况下,使用Real space进行计算可能会更容易收敛,并且可以避免由于过少的k点数导致的数值误差。
在一般情况下,当计算的原子数量超过200个,考虑到收敛性和计算资源的问题,通常建议使用Real space进行计算。但这只是一个经验值,具体情况还需要根据实际情况进行综合评估和调整。
5. ELF、电荷密度分布、差分电荷密度,三者有什么区别?
A:ELF是指电子局域函数,是用来描述电子在分子中传播和分布的测量方法,可以用来描述化学键和分子中电荷分布的特征。
电荷密度分布是指物质中电子的分布情况,常常表示为电子密度ρ(r)。
差分电荷密度指的是不同空间位置或者不同化学物质之间电荷密度的差异,可以用来描述分子中化学键的性质以及分子间的相互作用。
因此,ELF可以用来描述化学键的性质,电荷密度分布可以用来描述分子中电子的分布情况,而电荷密度差则可以用来描述分子间的化学键性质和相互作用。
6. 解释一下计算零点能E_ZPE为什么把最后一列振动数据加和除以 2000?
A:零点能(Zero Point Energy,ZPE)是指物质在低能量状态时仍存在的能量。E_ZPE是指零点能的能量,通常用于描述分子或原子系统的基态。它的具体计算式为:E_ZPE = 1/2 * ∑hω,其中h为普朗克常数,ω为振动频率。所以最后一列振动能数据加和除以2就获得了E_ZPE,再除以1000是用以单位换算。
7. 计算吸附结构的中心声子为什么要固定板层?
A:因为声子频率与晶格的局部振动方式密切相关,当吸附分子与板层相互作用时,其对板层原子的位置造成了一定扰动,从而影响了板层原子的振动模式。如果不固定板层,当计算声子频率时,板层原子在受到吸附分子的扰动作用下会发生移动,就无法准确地反映出吸附结构中心声子的振动情况。


