
利用Schrödinger(薛定谔)自由能微扰计算,预测蛋白-蛋白复合物突变的相对结合能。
基于自由能的计算方法,有望显著提升蛋白质设计的通量并降低成本。不过需要注意的是,此类方法必须达到很高的可靠性、准确性和自动化水平,才能有效应用于实际的工业场景,进而推动蛋白质设计项目的开展。
来自Schrödinger、阿斯利康和哥伦比亚大学的研究人员选取了文献报道的多种体系,针对单点突变导致的蛋白-蛋白结合亲和力相对变化展开了基准测试研究,所用方法正是自由能微扰(FEP+)计算。
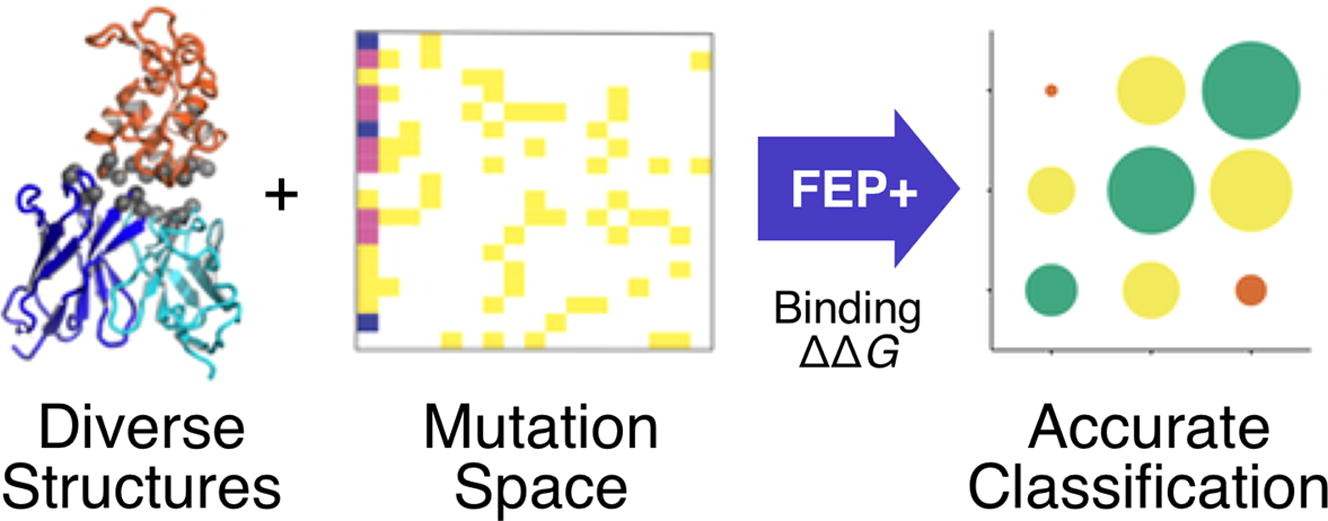
图注:研究摘要图
研究亮点
对多数蛋白质突变的相对结合自由能变化计算精度可达约1 kcal/mol。
采用Protein FEP+ Groups方法,自动化处理可滴定残基的交替质子化状态。
将FEP+方法成功应用于实际的蛋白质设计项目。
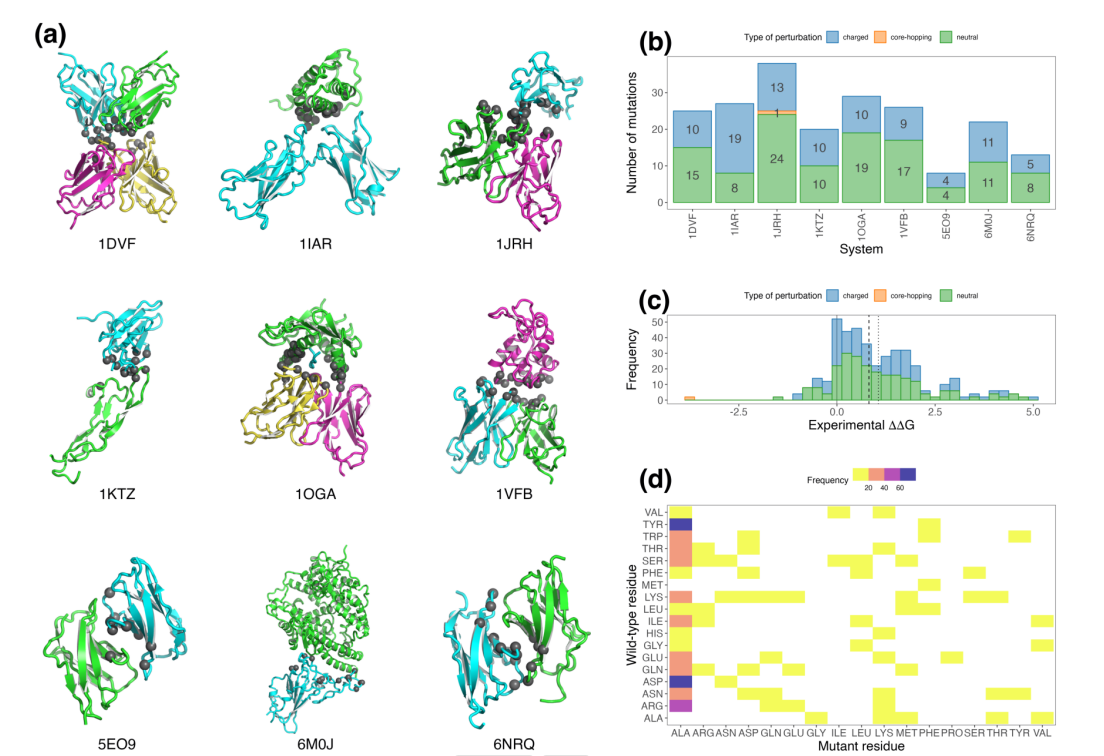
图注:基准数据集的体系和突变总览
这项研究提出了一种针对可滴定氨基酸交替质子化状态的稳健处理方法(称为“Protein FEP+ Groups”),与实验测得的结合自由能相比,该方法能提升相关性并降低误差(如下图)。用其处理pH值及不同质子化状态对结合作用的影响,可显著优化整体的计算结果。同时,基于FEP+计算模拟得到的化学、结构及能量特征,能够识别潜在的异常计算结果,且在一些情况下进行经验校正。
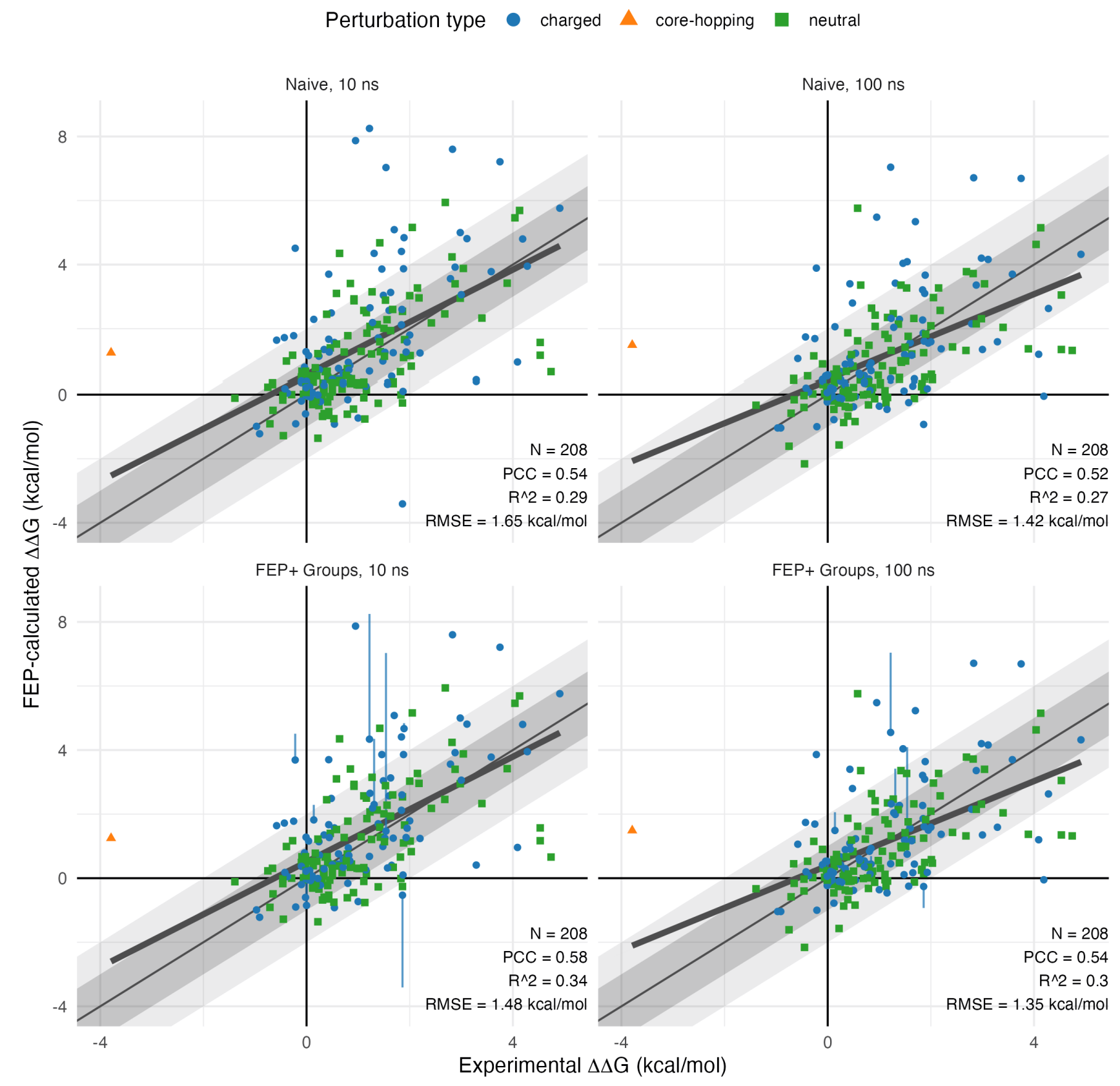
图注:FEP+结果与实验结果的对比
在完整的基准数据集中,部分误差较大的情况源于体系出现的构象重排现象(下图f)。这类现象既包括已观测到的情况(如1DVF wt中残基D:H33的构象发生改变,下图右),也包括可预判的情况(如表面环区的氨基酸突变为脯氨酸或由脯氨酸突变而来,或是小环肽中暴露于溶剂的极性残基突变为疏水性丙氨酸)。
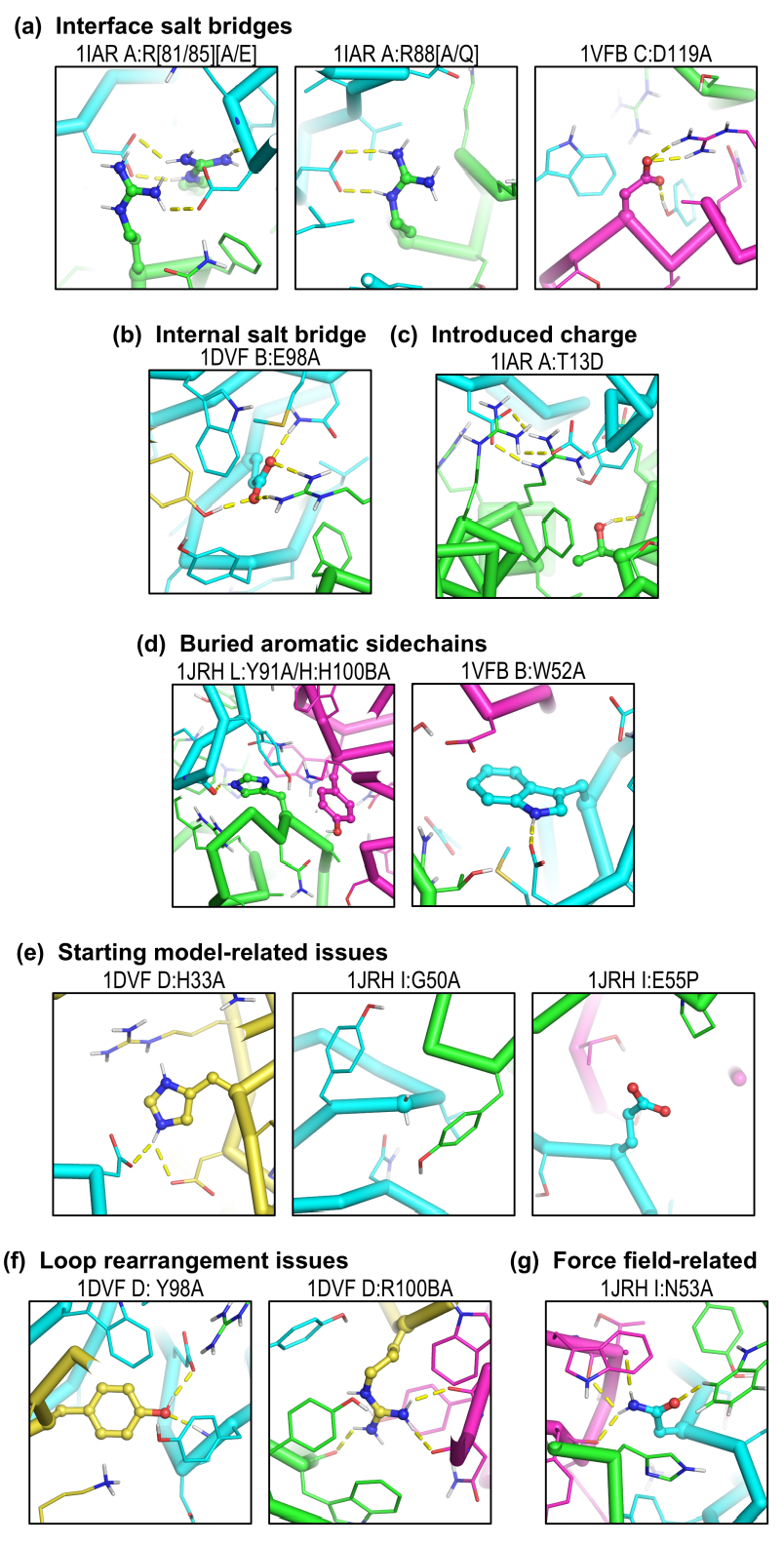
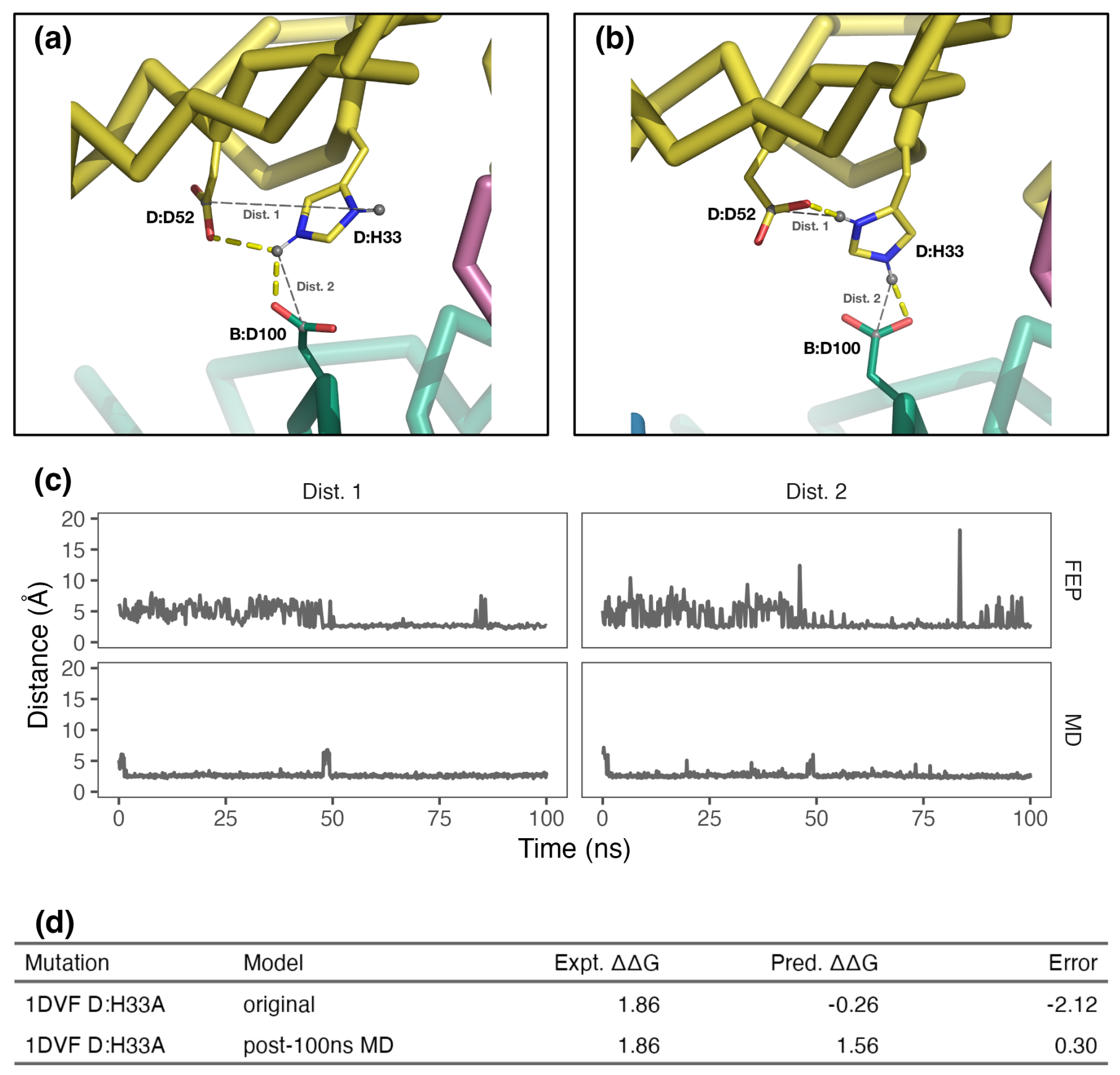
图注:绝对误差大于2 kcal/mol的异常情况类别(左)及1DVF D:H33A异常值分析(右)
通过对数据集中偏差最大的异常值进行细致分析,研究人员评估了默认FEP+计算流程的局限性,并开发了自动化脚本,能够识别可能需要进一步核查的潜在异常值,同时针对电荷相关的异常值计算经验校正值。
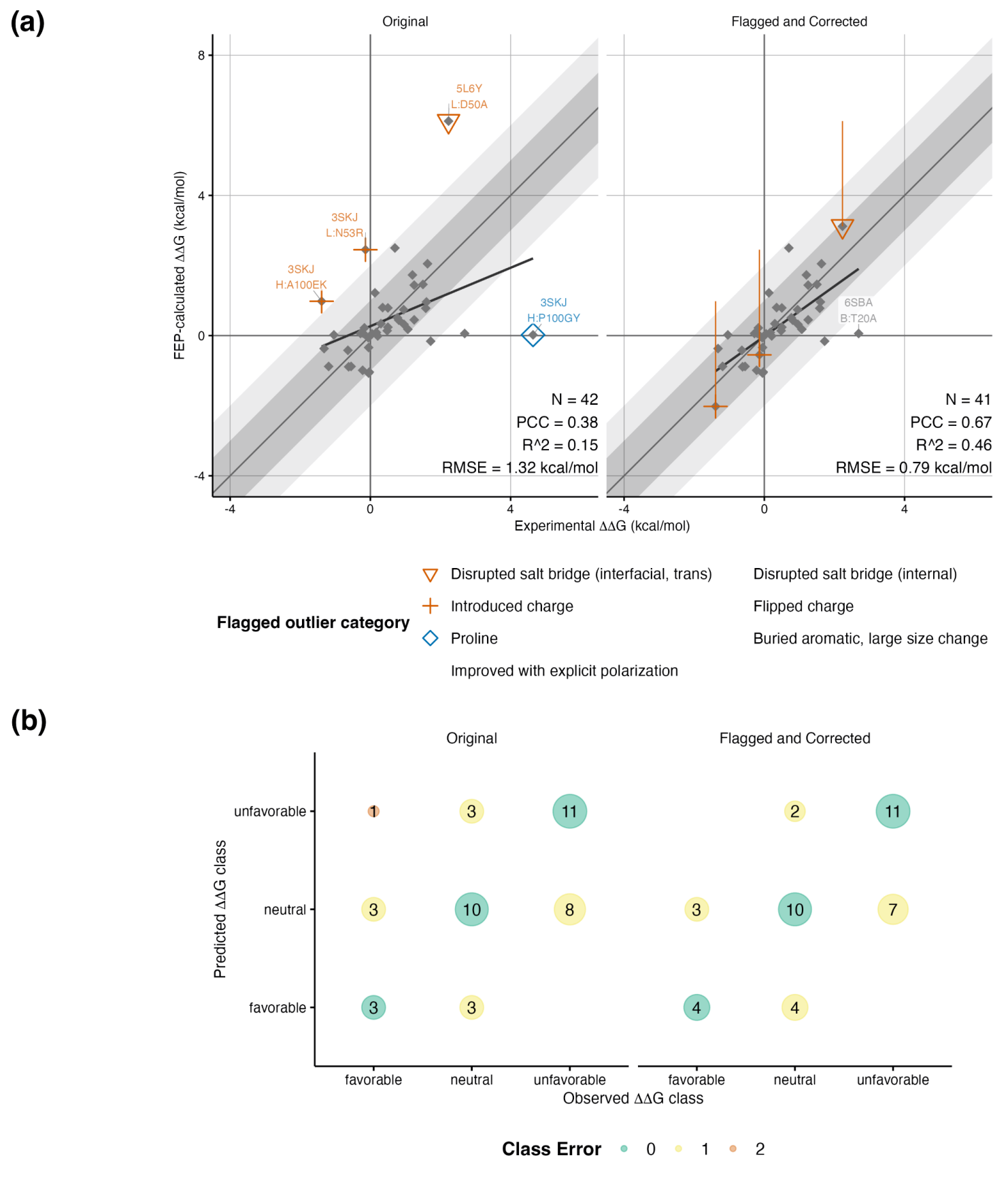
图注:基准数据集FEP+异常值的自动分类和经验校正
接下来,研究人员利用实际项目中的3个真实场景数据集,对上述异常值处理策略进行了验证(下图a)。数据集涵盖2种抗体-抗原体系与1种多肽-靶点体系的野生型及单残基突变体的表面等离子体共振(SPR)检测数据。
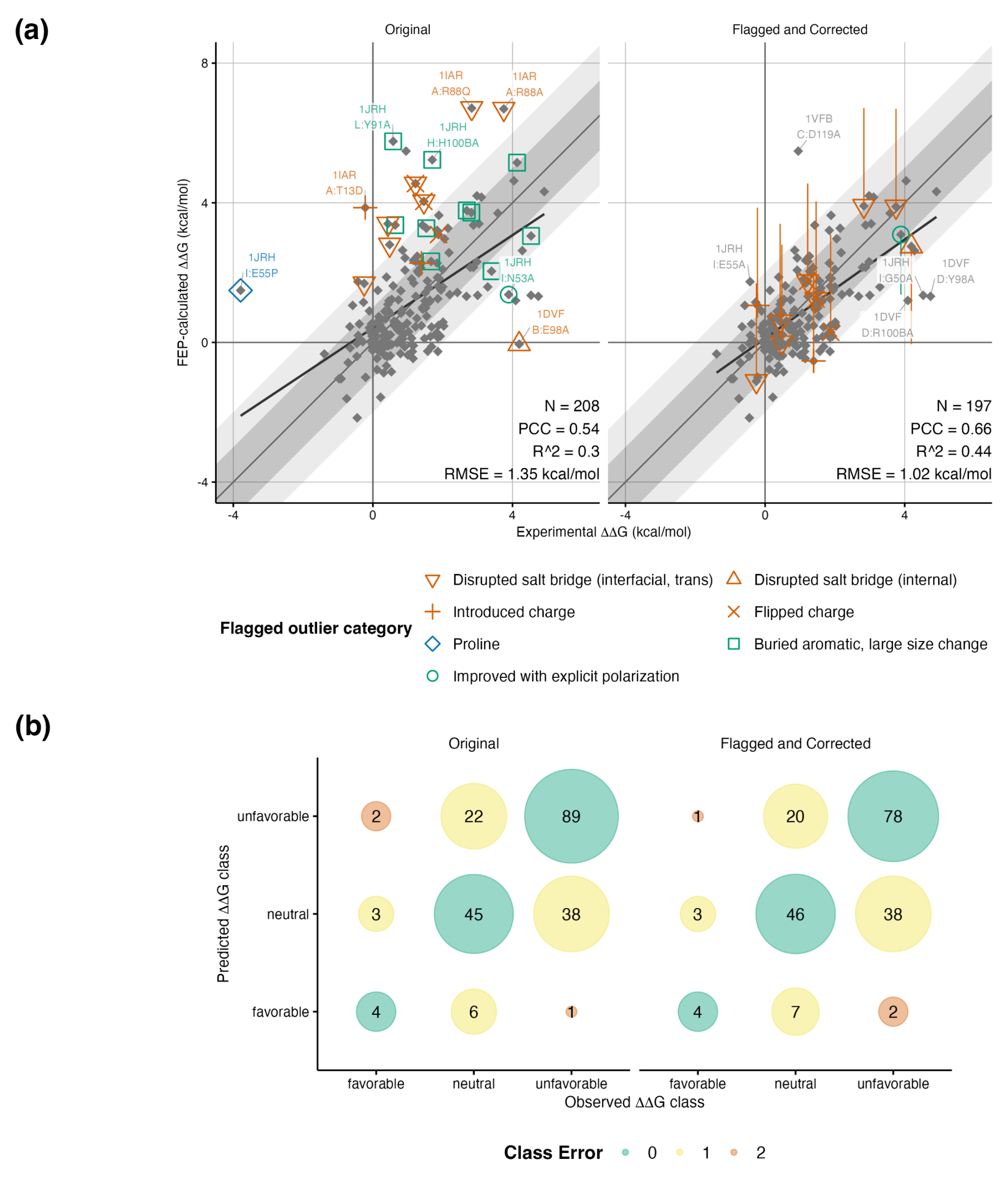
图注:自动异常值标记和校正在案例研究数据集中的应用(100纳秒的蛋白质结合FEP+计算)
结果显示,案例研究数据集的计算结果与基准数据集的结果相近,异常值自动标记与经验校正流程产生的效果同样与基准数据集的情况相近,在三元分类中取得了良好的准确率(上图b)。
总之,这项研究表明,对于多种体系中的绝大多数突变而言,蛋白质结合FEP+计算不仅能够得出准确的结果,还可指导生物制剂研发项目的实验环节。
欢迎联系我们获取Schrödinger软件试算!
参考资料
Sampson, Jared M., et al. "Robust Prediction of Relative Binding Energies for Protein–Protein Complex Mutations Using Free Energy Perturbation Calculations." Journal of Molecular Biology 436.16 (2024): 168640.


