
自由能微扰(FEP)计算是一种通过分子动力学模拟预测结合亲和力的方法。分子动力学模拟会考虑靶蛋白的构象动态变化,这使得FEP计算能够在兼顾诱导契合效应的同时,高精度筛选出强效化合物。之前的研究已经证实了FEP在设计强效共价型主蛋白酶(Mpro)抑制剂方面的显著效果。
近期,来自武田制药(Takeda)和Schrödinger(薛定谔软件)的科学家通过计算机辅助药物设计(CADD),发现了一类强效的非共价抑制剂,可抑制SARS-CoV-2 Mpro,且具有泛冠状病毒(pan-CoV)Mpro抑制活性,有望解决现有SARS-CoV-2治疗药物存在的各类局限性。

研究人员通过虚拟筛选识别出了一种以哌嗪为核心的非共价命中化合物。基于结构指导的骨架变形产生了一种新型三取代哌啶核心结构。在考虑Met49/Met165和Gln189诱导契合的条件下,通过FEP指导设计,成功筛选出强效的化合物30。该化合物不仅具有泛冠状病毒Mpro抑制活性,还对SARS-CoV-2奥密克戎变异株展现出细胞抗病毒效力。
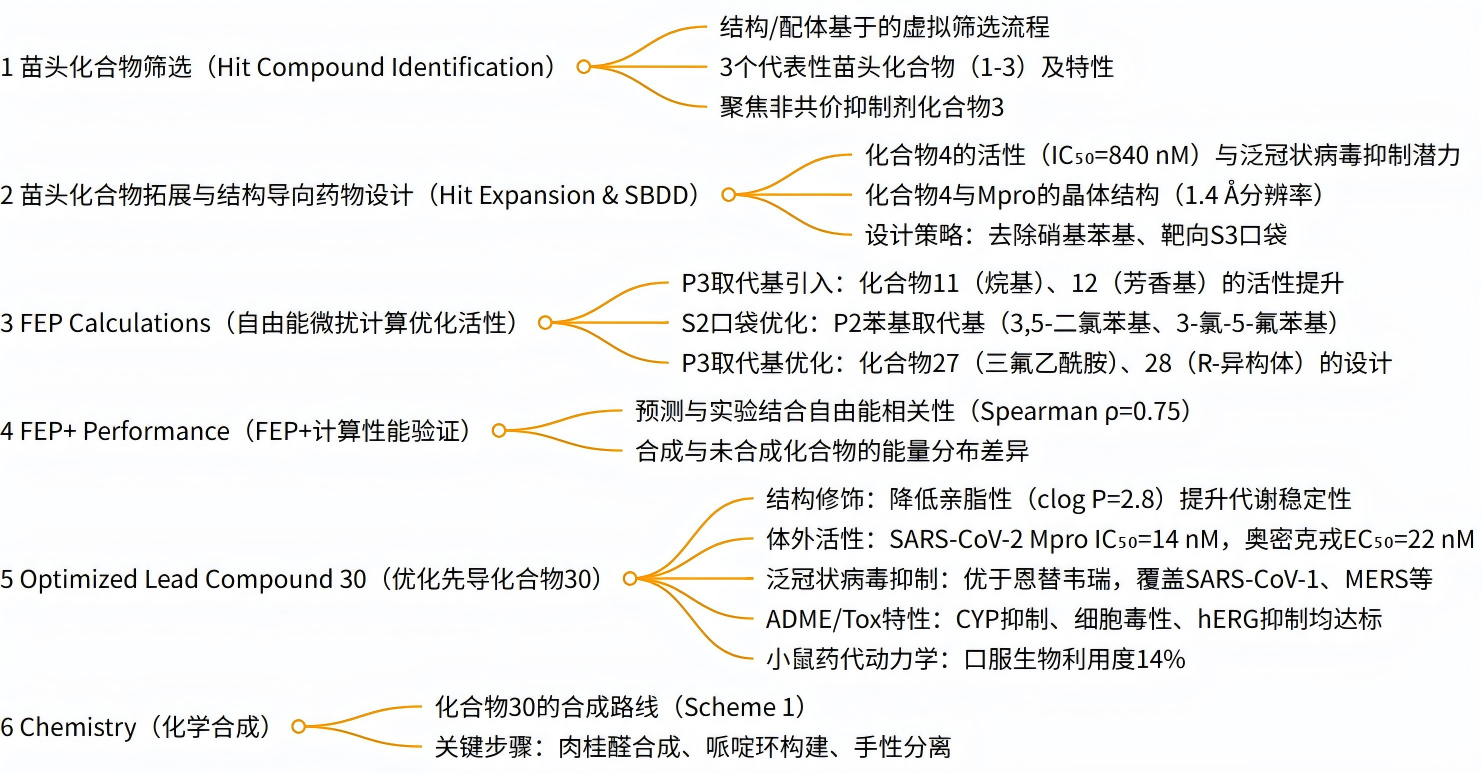
图注:研究的主要发现
1、苗头化合物虚拟筛选
虚拟筛选基于包含150万个化合物的武田化合物库。为构建包含所有可能异构体及电离状态的三维(3D)构象,使用Schrödinger分子模拟套件中的LigPrep工具,在pH值7.0±2.0的条件下基于OPLS2005力场完成构象生成。
筛选过程采用4个SARS-CoV-2 Mpro晶体结构作为对接模板。使用Schrödinger中Maestro模块的Protein Preparation Wizard,按默认设置对上述蛋白质结构进行预处理。随后,对这4个SARS-CoV-2 Mpro蛋白结构进行常规Glide SP对接,筛选非共价抑制剂。对SARS-CoV-2 Mpro晶体结构(PDB ID:6LU7)进行共价对接,筛选共价抑制剂。
除了上述基于对接的虚拟筛选外,研究人员还进行了基于配体的虚拟筛选。结合扩展连通性指纹(ECFP-4)、化学高级模板搜索(CATS)拓扑药效团模型及考虑原子/键/错配的最大公共子结构(MCS)搜索,从海量化合物中筛选出21个IC₅₀<30μM的苗头化合物(如下图A)。得到的非共价抑制剂化合物3,成为后续结构优化的关键起点。
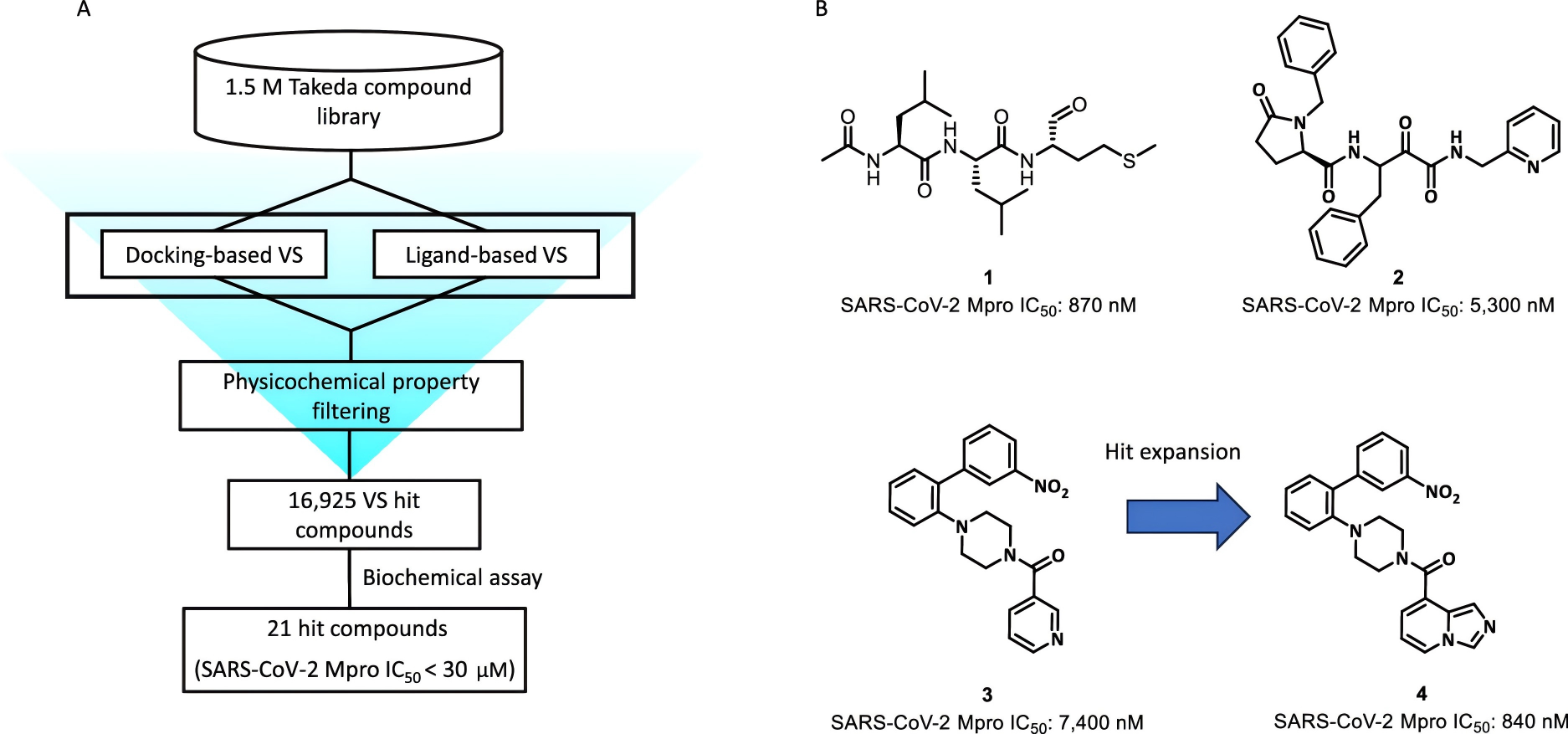
图注:(A)筛选苗头化合物的虚拟筛选流程。(B)代表性苗头化合物。
2、化合物优化
在结构导向的药物设计中,Schrödinger的FEP+计算发挥了核心作用,实现了化合物活性的定向优化。
针对S3口袋,为评估在哌啶核心引入P3取代基的效果,使用了SARS-CoV-2 Mpro与化合物4形成复合物的晶体结构,并在Maestro中手动绘制哌啶核心上的P3取代基,将其作为FEP+计算的初始结构
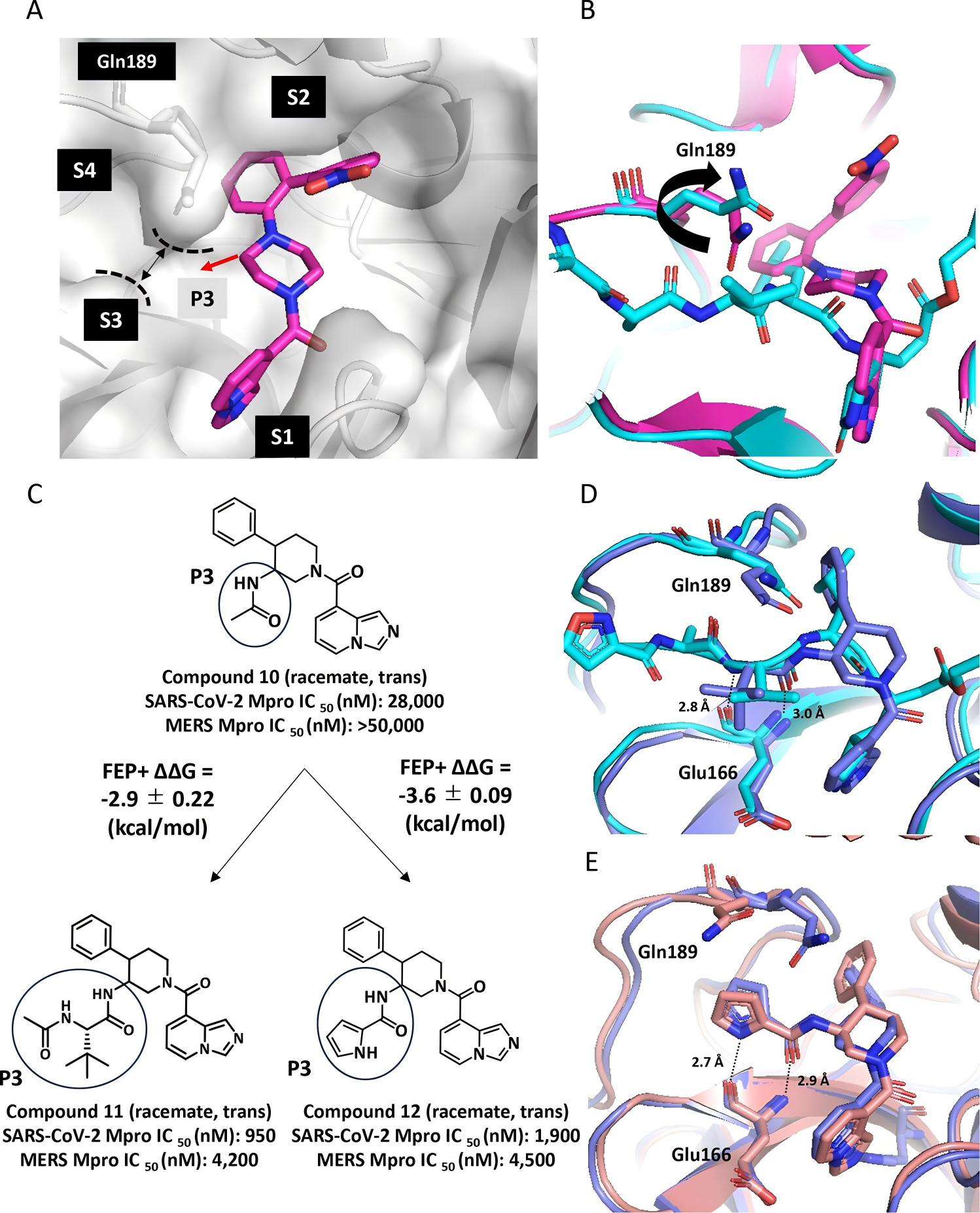
图注:FEP+计算预测P3取代基的效果
为探索苯基取代基以优化S2口袋相互作用,采用了SARS-CoV-2 Mpro与化合物12形成复合物的晶体结构,在Maestro中手动绘制苯环上的P2取代基,作为计算初始结构。FEP+计算设计出烷基取代的化合物11与芳香基取代的化合物12。
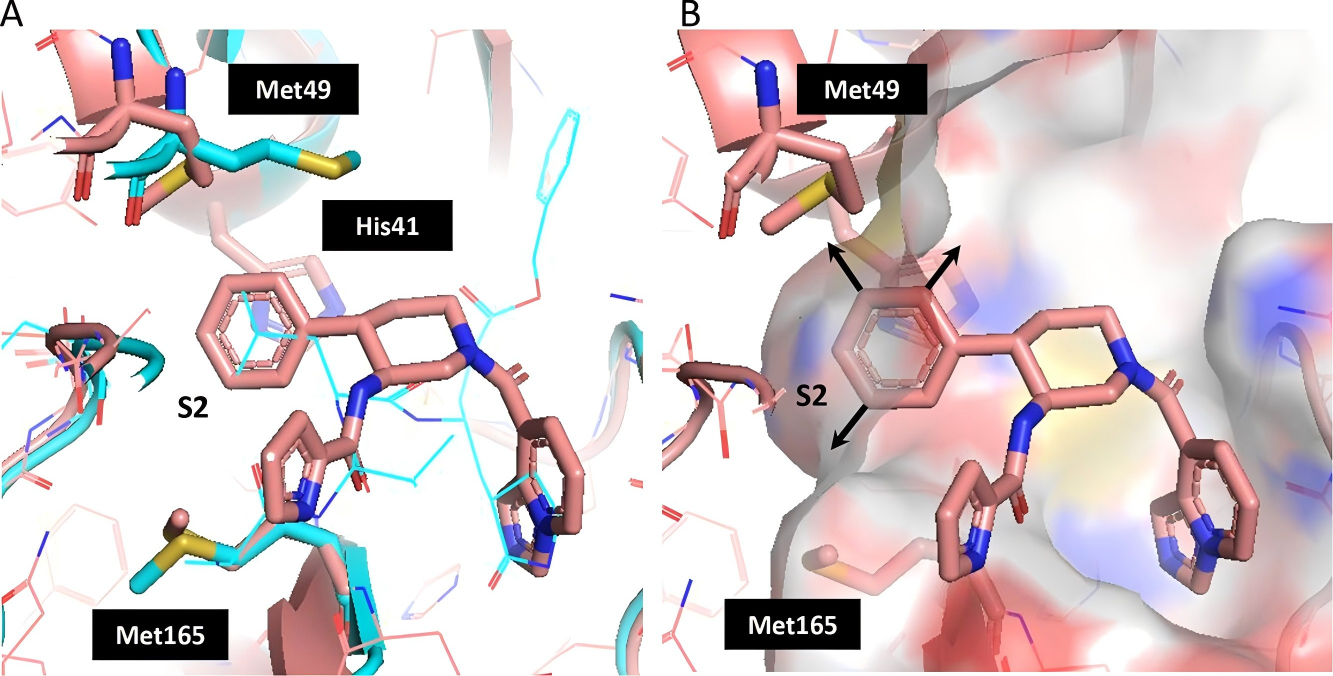
图注:FEP+计算预测P2取代基的效果
此外,FEP+还预测出P3-R异构体化合物28优于S异构体化合物27,且通过降低亲脂性(clogP从3.9降至2.8),助力优化得到代谢稳定性提升的化合物30。
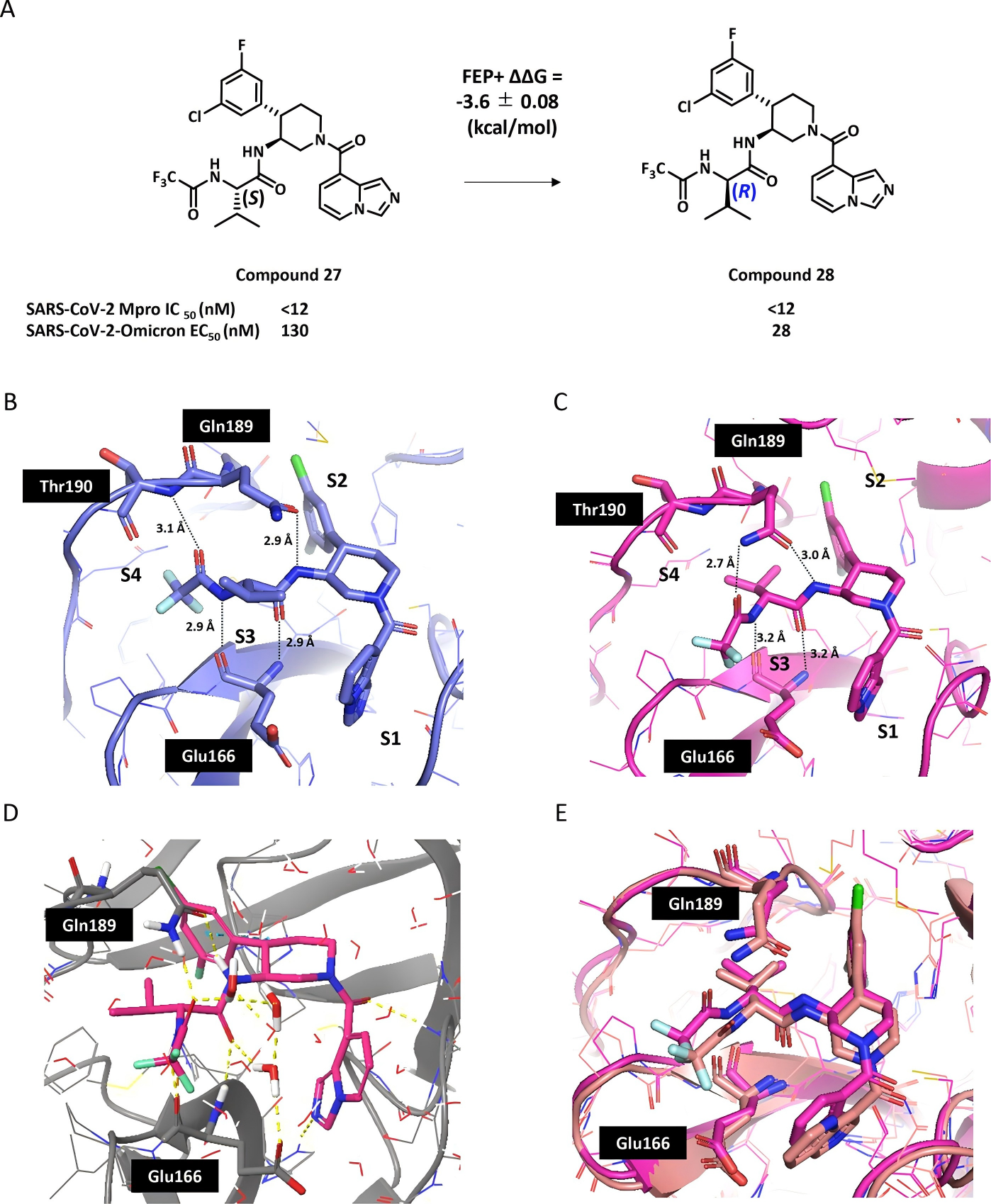
图注:化合物27、化合物28的结合模式与构象验证
3、性能验证
研究人员通过大规模FEP+计算进行了性能验证,7330个化合物通过LiveDesign自动运行,计算以SARS-CoV-2 Mpro与化合物12的复合物晶体结构为参照,通过Glide SP进行核心约束对接,生成输入构象(pose)。85个化合物进入合成阶段。
下图显示,FEP+预测结合自由能与实验值相关性显著(Spearman ρ=0.75,p<0.0001),且能有效区分合成与未合成化合物的能量分布(p<0.0001)。
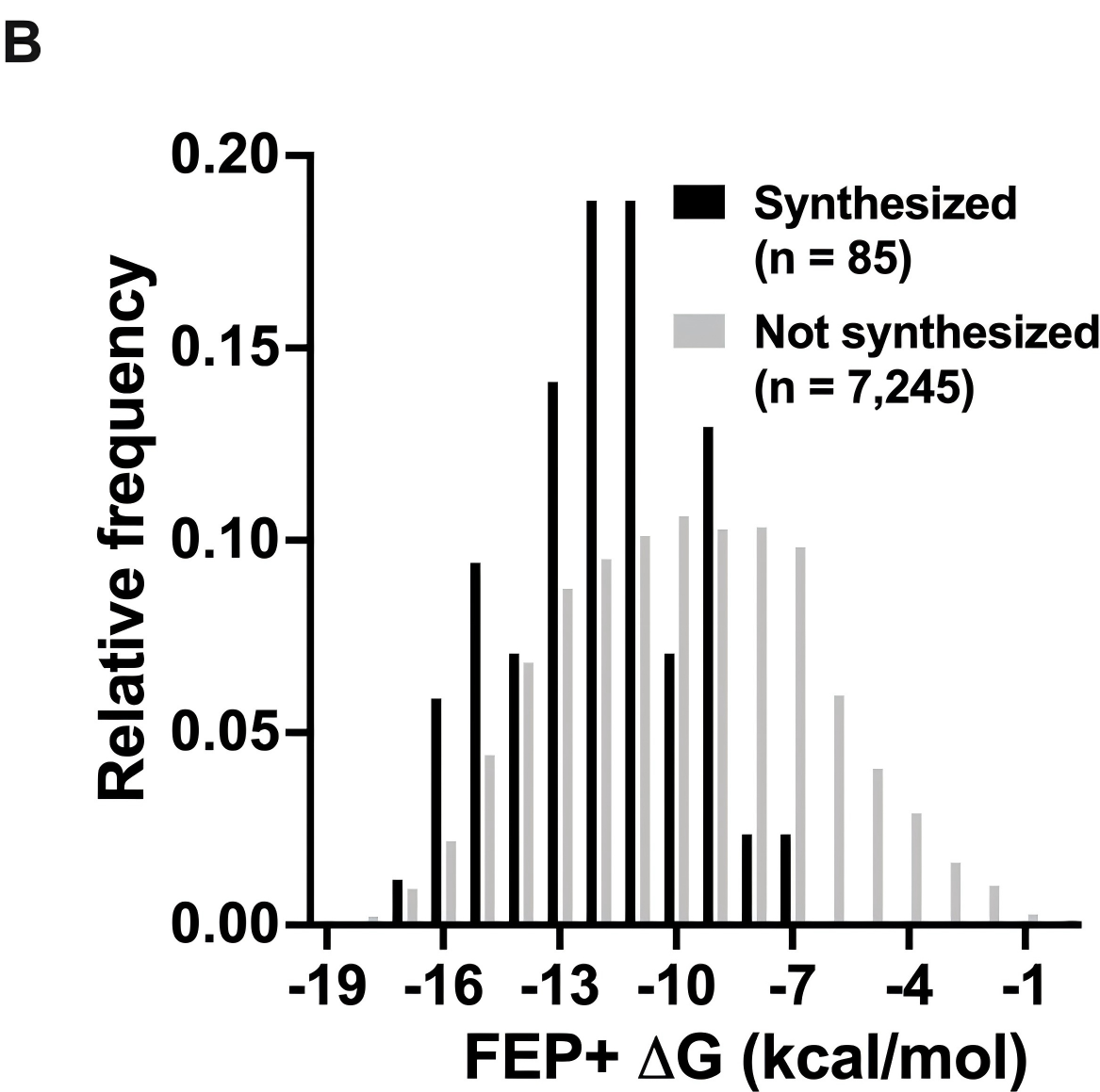
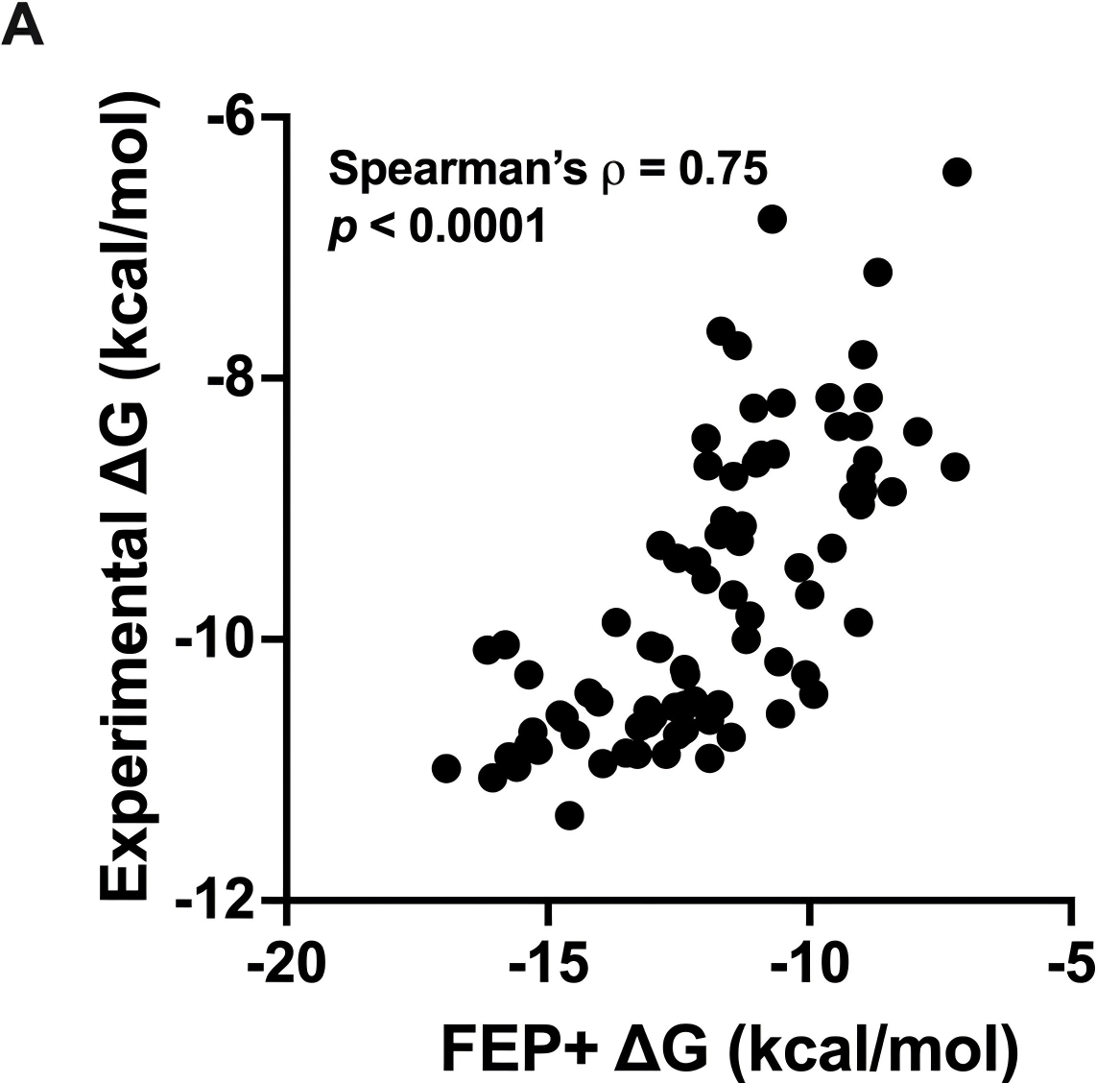
图注:(A)FEP+计算的前瞻性性能。(B)已合成化合物与未合成化合物的FEP+预测结合自由能分布。
这些结果证明了LigPrep、Glide及FEP+等工具在冠状病毒抑制剂设计中的适用性,为化合物30的强效活性(下图显示SARS-CoV-2 Mpro IC₅₀=14nM)提供了关键保障,彰显了计算化学在药物研发中的精准性与高效性。
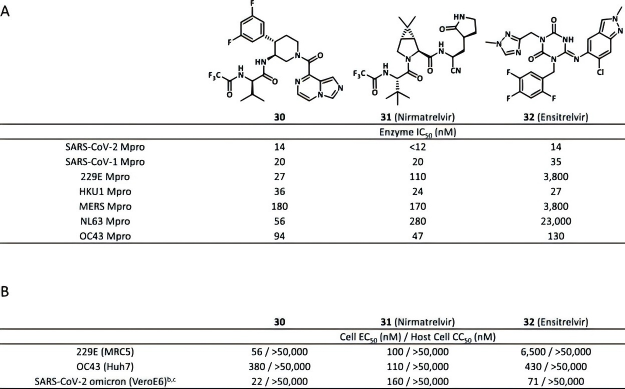
图注:(A)体外酶抑制活性,(B)体外细胞抗病毒活性。
在对先导化合物30完成优化后,研究人员通过体外药物吸收、分布、代谢、排泄与毒性(ADME/Tox)实验,以及体内小鼠药代动力学研究对其进行了表征。研究结果表明,化合物30有望成为助力未来大流行防控的有力工具。

参考文献
Okabe, Atsutoshi, et al. "Discovery of Highly Potent Noncovalent Inhibitors of SARS-CoV-2 Main Protease through Computer-Aided Drug Design." Journal of Medicinal Chemistry (2025).


