
CO2在铁改性二硫化钼催化剂上的吸附和解离:DFT研究
关键词:CO2还原,密度泛函,吸附,直接解离,MoS2,铁
1.案例背景
大气中温室气体(CHA)浓度的升高导致全球变暖,气候变暖、降雨量增加、海平面上升、臭氧层破坏和海洋酸化。二氧化碳(CO2)则是重要的温室气体,占总排放量的77%以上,但同时CO2又是一种有价值的化学原料。将CO2转化为有价值的碳产品被认为是,解决这些环境问题的有发展的战略。其中CO2直接解离成为CO,是CO2还原的关键步骤。CO不仅是有价值的化学品,CO2能够通过生成CO中间体进而还原成甲醇(CH3OH),甲烷(CH4)等。MoS2是一种结构类似于石墨烯的非贵金属基二维材料催化剂。因MoS2特殊的电子特性、可调特性、高的表面体积比和优异的热稳定性,成为了CO2和其它CHA非极性气体的高效吸附剂,并有望成为CO2还原催化剂。但是由于MoS2的活性位点密度低,电子输运效率低,以及导带不够负等问题,限制了MoS2的催化活性。因此本案例通过Fe负载和掺杂改性MoS2,通过DFT计算,比较了单个Fe和两个Fe改性的MoS2的CO2吸附能力和结构电子性质,发现带有硫(S)空位的双Fe原子掺杂的MoS2催化还原CO2能力最强。
2.建模与计算方法
作者通过MedeA InfoMaticA 搜索了MoS2晶胞。使用Supercell Builder 将MoS2扩展成4×4×1的超晶胞;使用Surface Builder切面得到MoS2(001)超胞表面;随后在MedeA Environment 中通过取代掺杂,构造了单S空位MoS2(S1V-MoS2)、单Fe原子负载的MoS2 (Fe1Dep-MoS2)、单Fe原子掺杂的MoS2 (Fe1Dop-MoS2)、双Fe掺杂的MoS2(Fe2DopS-MoS2和Fe2DopD-MoS2);随后采用MedeA-VASP模块中的DFT方法对各结构进行优化,同时计算各结构上CO2的吸附及活化,同时计算并分析了Bader、态密度、差分电荷密度等。截断能选取500 eV,K点基矢选取3×3×1, 计算过程考虑范德华作用力,采用 DFT-D3 计算。通过DFT计算得到了Fe改性MoS2的方法,最后通过MedeA-TSS模块研究了CO2在不同方法Fe改性的MoS2上直接裂解的反应能垒,为后续MoS2基催化剂研究打好了基础。
3.结果与讨论
3.1 催化剂结构和CO2的吸附:
表1 结合能(Eb,eV),磁矩((M, μB),Fe原子上的电荷(Q, e),单和双Fe原子MoS2的电荷转移(Δq)
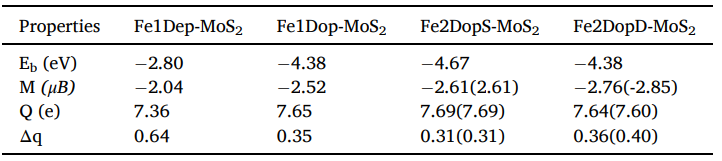
本案例中,在优化好各结构后,作者首先计算了各结构的结合能,判断了改性后的结构稳定性,如表1所示,结合能越大代表结构越稳定。其结合能排序为Fe2DopS-MoS2> Fe2DopD-MoS2 ≈ Fe1Dop-MoS2 > Fe1Dep-MoS2。结果表明两Fe原子掺杂在相同Mo原子上的MoS2结构更稳定。随后通过第一性原理分子动力学(AIMD)计算,发现Fe改性催化剂的键只在小范围振荡,验证了Fe掺杂结构稳定性的特点。另外,研究表1可以发现,Fe改性MoS2后,Fe上富正电荷,也就是说Fe原子为电子给体,可作为CO2吸附活性位点。
CO2直接解离的第一步是在催化剂表面吸附CO2。为了在催化剂表面获得最稳定的CO2吸附结构,考虑了六种MoS2催化剂上CO2吸附的不同结构,经结构优化后得到了能量最低吸附络合物,如图1所示,计算了各催化剂的吸附能(如图2)。吸附能越负代表CO2吸附性越强,其吸附能力强弱遵循:Fe2DopS-MoS2 > Fe2DopD-MoS2 > Fe1Dop-MoS2 > Fe1Dep-MoS2 >S1V-MoS2 > pristine MoS2。pristine MoS2和S1V-MoS2与CO2的吸附为范德华力作用,属于物理吸附,故吸附能较小。MoS2经Fe原子掺杂和负载之后,CO2吸附能力明显上升,这是因为CO2中的O原子倾向与催化剂中Fe原子成键,为化学吸附,且吸附距离更短。其中,两个Fe掺杂的Fe2DopS-MoS2的CO2吸附能力最强,且吸附后直线型的CO2键角发生弯曲,键角从180°变为142.21度,这说明Fe2DopS-MoS2为潜在的CO2吸附材料。
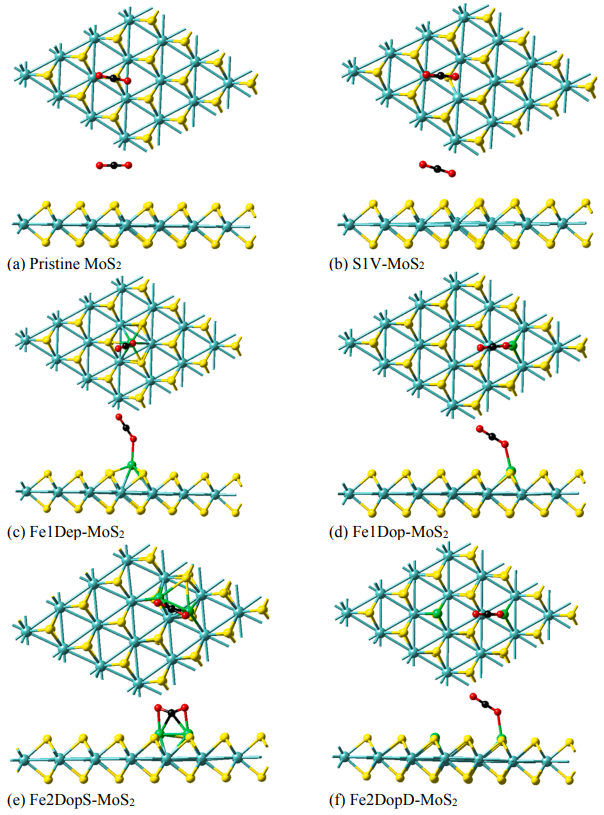
图1 优化后的单S空位MoS2(S1V-MoS2)、单Fe原子负载的MoS2 (Fe1Dep-MoS2)、单Fe原子掺杂的MoS2 (Fe1Dop-MoS2)、双Fe掺杂的MoS2(Fe2DopS-MoS2和Fe2DopD-MoS2)的俯视和侧视图。
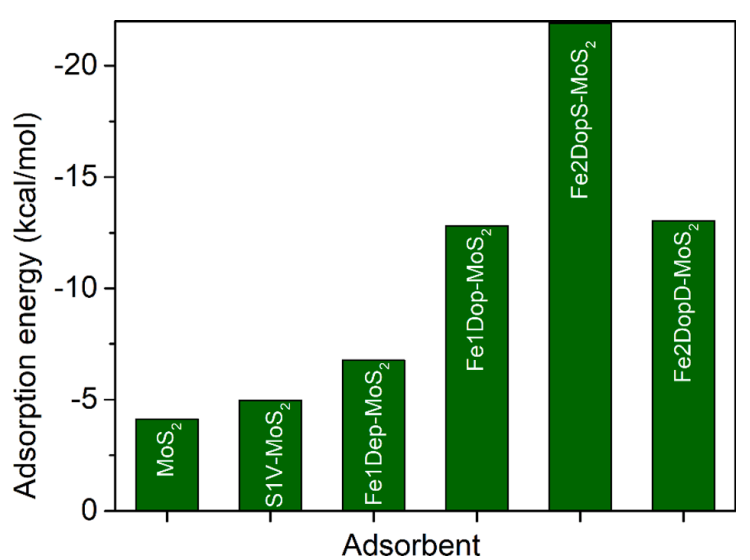
图2六种MoS2催化剂的CO2吸附能。
而后作者计算了六种催化剂吸附CO2后的电子性质。图3比较了六种催化剂吸附CO2前后的态密度(DOS)。六种催化剂在吸附CO2后,只有Fe2DopS-MoS2的DOS发生显著的偏移,并在费米能级附近产生新的峰,这个结果恰好与Fe2DopS-MoS2上CO2吸附能力最强的结果相对应,并且通过差分电荷密度(图3)分析得到Fe2DopS-MoS2上CO2得电子量最高,与催化剂电荷交互最强。
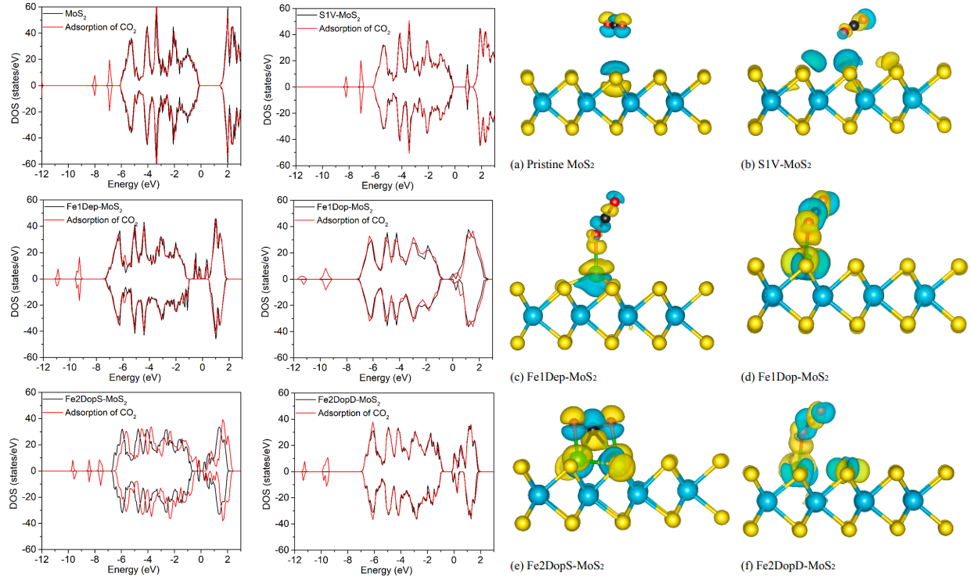
图3六种MoS2催化剂吸附CO2前后的态密度(DOS)和吸附CO2的差分电荷密度
3.2 CO2解离过渡态:
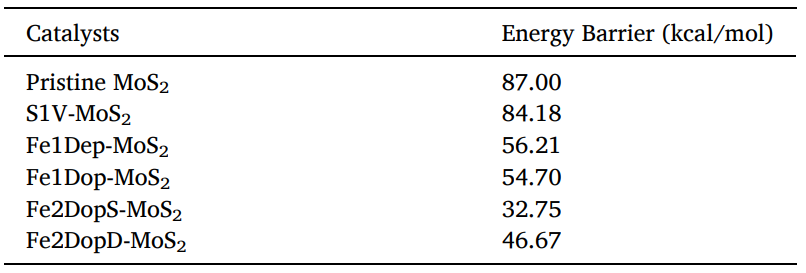
为了深入探究Fe改性后MoS2是否具有优异CO2裂解性能,作者使用MedeA-TSS搜索了六种催化剂上CO2裂解为CO的过渡态结构,如图4所示。图中所有过渡态结构中的CO2的C-O键长都延长了,并且O-C-O键角都是弯曲的。pristine MoS2 、S1V-MoS2、Fe1Dep-MoS2、Fe1Dop-MoS2、Fe2DopD-MoS2上CO2裂解的过渡态都是通过O原子与Fe原子相互作用,形成新的Fe-O键从而使得C-O键被拉长。而在Fe2DopS-MoS2上,CO2的C原子和O原子会分别与两个Fe原子发生相互作用,产生O-Fe键和两个C-Fe键。随后比较了六种催化剂上CO2裂解能垒(表2)。其大小顺序为:MoS2 > S1V-MoS2 > Fe1Dep-MoS2 > Fe1Dop-MoS2 > Fe2DopD-MoS2 > Fe2DopS-MoS2。结果显示,改性后的MoS2均在不同程度上增强了CO2裂解能力,其中Fe2DopS-MoS2上CO2的裂解能垒最小(32.75 kcal/mol),对比于未改性的MoS2(87.00 kcal/mol)能垒减小了54.25 kcal/mol。因此Fe原子的掺杂不仅使得MoS2对CO2的吸附能力增强,也改变了MoS2的电子结构,使其催化裂解CO2能力增强,其中两个Fe原子掺杂的Fe2DopS-MoS2是最优异的CO2裂解催化剂。
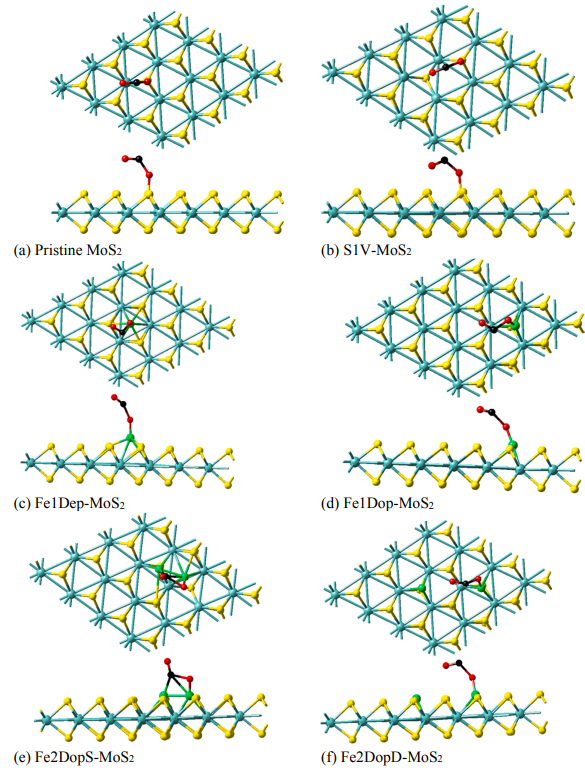
图4六种MoS2催化剂上CO2裂解为CO的过渡态
表2 CO2在六种催化剂上的裂解能垒
4.总结与展望
本案例中,作者系统的研究了CO2在单S空位MoS2(S1V-MoS2)、单Fe原子负载的MoS2 (Fe1Dep-MoS2)、单Fe原子掺杂的MoS2 (Fe1Dop-MoS2)、双Fe掺杂的MoS2(Fe2DopS-MoS2和Fe2DopD-MoS2)上的直接解离。与现有的CO2还原催化剂相比,本案例中的催化剂是经济可行的、环保的。作者计算了催化剂表面的CO2吸附以及CO2解离为CO的直接活化。获得了吸附能和反应势垒,以筛选出了最优异的催化剂。此外,还分析了Bader电荷转移、态密度以及电荷密度差异,探索其吸附CO2的电子结构。本案例为MoS2基催化剂的改性设计提供了指南。
参考文献:
C. Wu, W . Yang, et al. CO2 adsorption and dissociation on single and double iron atomic molybdenum disulfide catalysts: A DFT study [J], Fuel, 2021, 305: 121547.
使用MedeA模块:
MedeA Environment
MedeA-VASP
MedeA-TSS


