
前言
细胞色素P450超家族(CYP450)是一大类各种各样的酶,其主要功能是催化氧化有机化合物。细胞色素P450在内源性(如脂肪酸、类固醇、前列腺素和胆汁酸等)和外源性物质(如药物、环境致癌物和食品添加剂等)的代谢过程中,具有重要作用,而且是药物代谢与生物激活作用的主要酶类,约占到各种代谢反应总数的75%。细胞色素P450是由许多微粒体和线粒体亚铁血红素-硫酸盐蛋白组成的,广泛存在于细菌、真菌、植物和动物体内。
CYP1B1是细胞色素P450超家族的一个亚型,参与多环芳香烃等前致癌物的代谢活化,并在17-β-雌二醇诱导的乳腺癌发生与发展过程中起到了关键性作用。该酶在肿瘤组织中的特异性高表达及在肿瘤细胞耐药中的作用,也已被大量研究证实。该酶的特异性分布及在肿瘤发生与发展中的重要地位,使得它成为抗肿瘤药物研究中的新靶点。其抑制剂研究,在肿瘤预防及克服肿瘤耐药方面具有重要意义。
实验过程
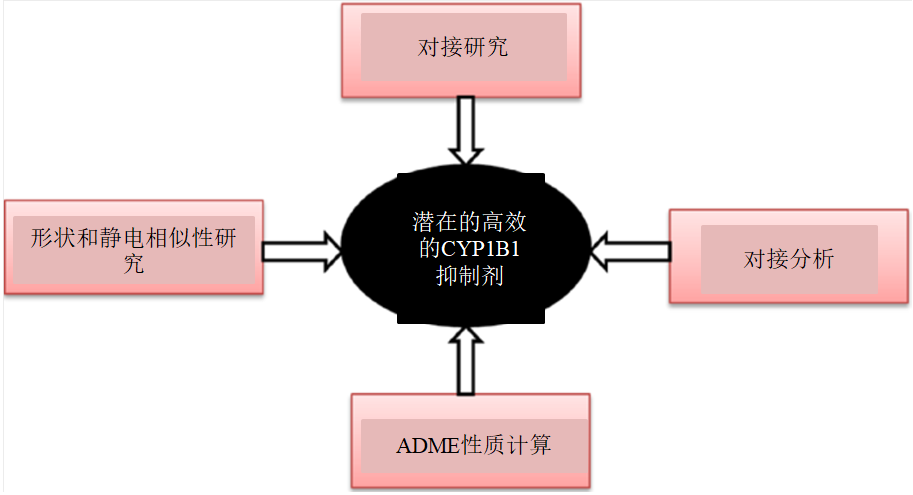
图1 实验思路
研究使用形状形似性方法(ROCS)、静电相似性方法(EON)、分子对接方法和ADME性质预测方法预测了新发现的化合物BNUA-3和BNUB-13的抑制活性。从计算机辅助的角度评价了新化合物的成药可能性和抑制活性,从而缩短了药物发现的时间和成本。
实验结果
1. 形状和静电相似性研究
图2为已知抑制剂和新化合物的结构。本研究使用ROCS和EON方法比较了新化合物BNUB-13和已知抑制剂TMS的形状相似性和静电相似性(如图3),相似性值分别为0.769和0.464。由两者的相似性结果可知,新化合物与已知抑制剂的3D相似性较大,可以有效地进入CYP1B1蛋白的活性口袋。
抑制剂TMS和ANF是CYP非选择性抑制剂,当结构中的桥链被脲基取代以后选择性会提高。因此化合物BNUB-13对CYP1B1的抑制活性IC50达到69 nM,选择性也高达62倍。结合已知抑制剂ANF和BNUB-13的结构特点,我们设计了化合物BNUA-3,保留了桥链结构中的脲基和平面性,结果发现抑制活性和选择性都得到了较大提高。并且实验结果和计算结果一致,这为新型特异性CYP1B1抑制剂的发现提供了指导。
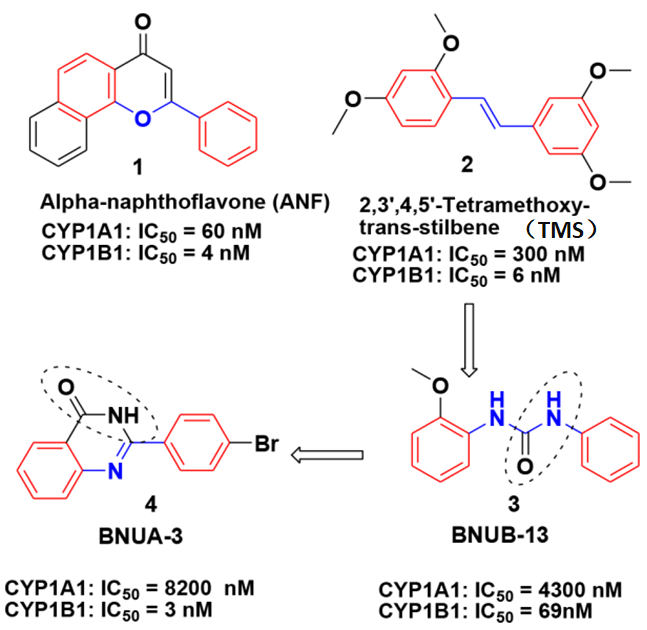
图2 已知抑制剂ANF和TMS,新化合物BNUA-3和BNUB-13的结构
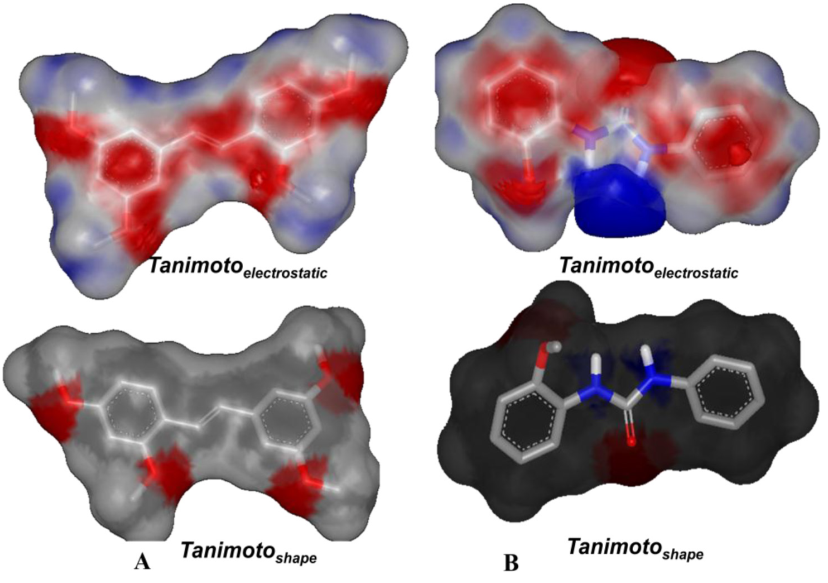
图3 抑制剂TMS(A)和新化合物BNUB-13(B)的静电和形状相似性图
2. 分子对接研究
从CYP1B1蛋白的X射线晶体结构中发现它的活性位点呈圆形窄缝状。当活性口袋被抑制剂ANF占据以后,会阻碍血红素铁-氧中间体与血红素的相互作用。对接研究发现,化合物BNUB-13与血红素铁-氧形成较强的范德华相互作用(< 5 Å)。脲基形成的芳香环与残基Phe231和Phe268形成π-π相互作用。化合物BNUB-13还与残基Asp333形成极性相互作用(图4C)。在对接研究中发现这些脲基化合物与CYP1B1蛋白形成了较强的相互作用,从而导致其抑制活性IC50较小。
化合物BNUA-3的芳香环C朝向血红素原子,含杂原子的B环与血红素形成范德华相互作用,该结合模式和已知抑制剂ANF相似。化合物BNUA-3的芳香环C上氧原子距离血红素的铁辅助因子最近,环A、环B与残基Phe231形成疏水相互作用。另外,化合物BNUA-3也与残基Phe134形成π-π相互作用。这些相互作用是导致化合物产生抑制活性的重要因素。本研究涉及到的4个化合物都较好的占据了圆形窄缝状的活性口袋,并且与残基Phe231、Phe268形成相互作用,芳香环结构与血红素形成较好的相互作用。不同之处在于,化合物BNUB-13可以与残基Asp333形成极性相互作用,BNUA-3可以与残基Phe134形成疏水作用,并且化合物BNUA-3芳香环C上氧原子与血红素的铁原子距离很近。相互作用的不同之处是导致化合物的抑制活性和选择性提高的原因。
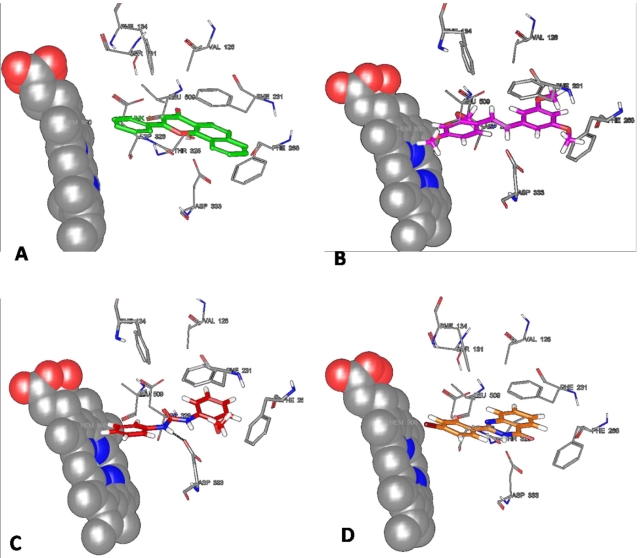
图4 抑制剂ANF(A)和TMS(B)、化合物BNUB-13(C)和BNUA-3(D)与CYP1B1蛋白的相互作用模式(PDB ID:3PM0)
3. ADME性质预测
使用计算机辅助的方法计算了化合物的偶极矩、口服生物利用度和血脑屏障穿透性(PlogBB)。预测结果表明,新化合物符合类药五规则:氢键受体(HBA)不小于10;分子量(MW)不超过500 D;氢键供体(HBD)小于5;油水分配系数小于5。血脑屏障穿透性(PlogBB)的预测结果表明没有一个分子能穿过血脑屏障对神经系统造成毒性。
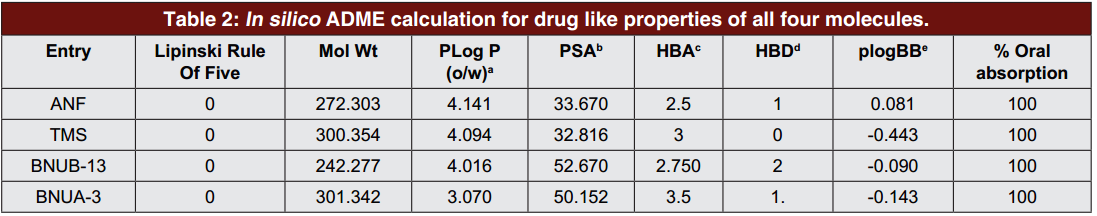
图5 ADME性质预测结果
结论
配体和蛋白活性口袋之间的相互作用在药物设计和发现中扮演着重要的角色。通过比较和计算已知抑制剂的形状和静电性质为新化合物的设计提供重要的指导。本研究对已知抑制剂和新化合物的形状、静电性质进行了比对,发现两者有相同的性质。该结果从计算角度表明新化合物可以作为CYP1B1蛋白抑制剂,这和实验测得的活性结果IC50一致。ADME性质计算结果发现这两个新化合物符合成药规则。


