
简介:
现代生物研究中的高通量技术如microarray、蛋白质组学或NGS能够让科学家们检测到几乎所有的mRNA,蛋白质或DNA序列的变异,从而获得成千上万的数据。分析数据结果的复杂程度和所需要的时间也随之直线上升,科学家们往往会陷入如何从海量的实验数据中挖掘到该体系到底发生了什么的泥潭中。想充分挖掘实验数据中的价值,需要科学家多方面的知识和技能,既要从生物学角度去阐释整个实验系统,又要理解系统变化的原因和效应等。科学家们通常只去寻找实验数据中发生差异表达的基因的上游调控子,如转录因子或调控的microRNA。但要完全理解实验结果的效应,科学家们必须进一步分析差异基因所调控的分子通路,生物学功能,已知的毒理学效应并对某些特定的关键分子进行进一步的全面调研(i.e. 后续靶标或生物标志物)。
以前科学家们可以依赖于个人的生物学专业知识并辅以检索最新文献来进行简单的数据分析。但随着文献的研究领域分类更加细化,知识的积累和文献的调研变得不再那么简单。现在,科学家们开始使用基于互联网的软件工具,包括专业的网站(i.e. PubMed)和一些免费的或商业化的分析工具(i.e. DAVID, Ingenuity-IPA)来帮助收集并分析数据。常规的高通量数据的分析和进一步的实验假设,一般均从阅读尽可能多的相关文献并调研实验结果中变化最大的基因开始。然而这样的分析策略往往会大量遗漏关键的信息,很多时候是因为相关的分子数据库和实验数据相关的文献量非常大,以至于科学家们无法面面俱到。而二代测序(NGS,如RNA-sequencing)的数据相当于为microarray实验提供了更加准确的转录本和同源基因信息,使获得信息变得更加复杂。因此,能够深度挖掘实验数据、将各种来源的背景信息整合在一起并提供灵活易用的工具进行查询的软件对理解实验结果变的日益重要。
在本文中,我们比较了商业化软件IPA和数种免费的常用组学数据分析软件在高通量数据分析的速度和效果。为了比较得出不同软件的分析能力和效率,我们设计了三类代表性的分析任务:1. 查询一个不熟悉的基因,建立实验结果的生物学假设;2. 分析基因表达谱数据(转录因子,通路和生物学效应);3. 鉴别被microRNA所调控的靶标基因。
实验一:查询一个不熟悉的基因,建立实验结果的生物学假设
本实验中,我们以四个有潜在治疗意义的靶基因为例进行搜索:膜联蛋白2(annexin2 ANXA2),瘦素受体(leptin receptor LEPR),基质淋巴细胞生成素(TSLP)和白介素13(IL-13)。对比了三个工具:IPA,NCBI(Entrez),Pubmed在做这几个基因搜索时所用的时间。其中,IPA的搜索速度快速,获得的信息较为全面和准确。Pubmed所需时间比IPA略长,NCBI Entrez搜索最慢。对ANXA2的搜索结果均不太准确,IPA耗时约1.3小时,而NCBI Entrez则需要8小时之久。对于在肥胖和心血管研究中被受关注的LEPR,IPA大约需要3.24小时,而NCBI Entrez需要86.3小时。获取信息的方便程度由搜索目标的格式不同而不同(图一)。
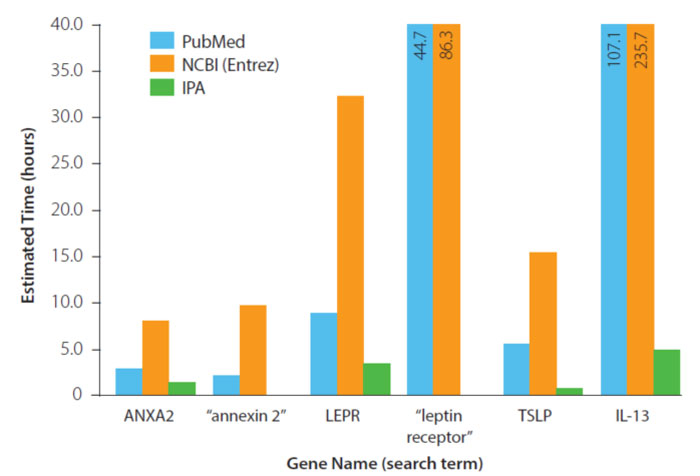
图一:寻找一个基因所需的时间。计算时间的方法是根据每个搜索中找到的文献和其他信息的数量而定的(阅读资料所需的时间)。在同义词搜索时由于用不同的搜索关键词而产生了多种搜索结果。图中。坐标最大值为40个小时,超出这个范围的数据直接标记在图中。
实验二:使用成熟的分析软件分析生物芯片和RNA-SEQ基因表达数据的数据
本实验将用分析一个例子所需的时间作为衡量的标准,将从数据上传开始计时一直到获得一个确定的结果比如信号通路或生物学作用为止定义为初级结果时间(TTFR)。我们对比了Cytoscape,DAVID,GenMAPP,IPA和Path Visio几个成熟分析软件对表达谱芯片数据分析出潜在的转录因子,生物通路和生物学作用等结果所需要的时间。
几种工具的准备工作所需要的时间各不相同,DAVID,IPA和Path Visio几乎不用什么时间准备,GenMaPP需要预先下载好相应的基因注释数据库,而Cytoscape需要预先下载一个插件,并且要人工导入生物通路和相互作用数据库(比如使用插件调用PathwayCommons的数据)。
表一为使用这几种方式分析高通量数据时所需要的时间。++代表了获得了可视化的结果和预测了可能的影响。+只能给出基本的基因通路,无针对性。+/-表明只能给出不完全的结果。N/A表示不能完成类似的工作。
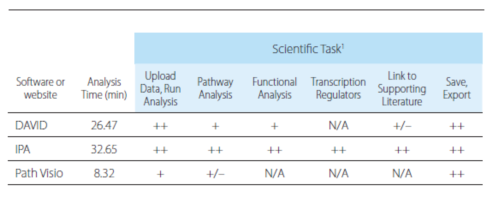
本实验中,我们主要对比了DAVID和IPA在分析数据上的差别。IPA上传数据非常灵活,可以由用户自定义数据的类型和需要分析的数据列。IPA和DAVID分析得到初级结果的时间(TTFR)差不多,都是30分钟左右。只是IPA是由上传的实验数据为根据分析其中差异显著的基因的生物通路和生物学作用,而DAVID给出的生物通路与上传数据中显著差异基因完全没关系。
IPA在短短的30分钟内就能从表达芯片数据中差异显著的基因根据预定的8种对比条件下挖掘出其生物学通路,生物作用,转录调节因子,以及相应的文献支持等分析结果(表一,图二)。而DAVID虽然也是30分钟能完成初级分析,但是其一次只能分析一种对比条件。不同的条件均要分别输入分析,这样就很难直接对比不同条件的下的分析结果(表一,图二)。

图二:基于不同组病人基因表达值用IPA预测生物学作用。由z-score做出的热图是基于基因表达显著差异和实验中基因的预期效果所确定的。A)对治疗有应答的病人基因表达显示参与炎症反应的基因都下调了,而细胞凋亡的基因有些上调有些下调。B)对治疗无应答的病人基因表达中,参与细胞凋亡的基因都上调了。C)表格中显示参与嗜酸性细胞的迁徙的基因表达有上升趋势(z=1.683)
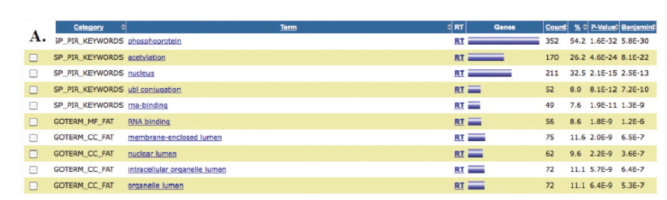
图三:由DAVID做出的分析结果。A)根据InterPro和GO定义出的功能注释柱状图。B)KEGG中脂肪酸代谢的通路图(部分)红色的星表示出该基因来自上传的数据,绿色的填充色表示小鼠同源体。
实验三:对MicroRNA-mRNA靶向作用的关系预测
对microRNA调节基因表达的研究近些年来非常热门。由之产生的出来一些列的数据库和工具用于分析和预测microRNA的靶向,比如TargetScan和PicTar。但是由于一条microRNA可以由于所处的组织,疾病,通路的不同能够靶向上千条目标基因。于是,对其靶向的准确分析将为之后的验证试验节约大量成本。
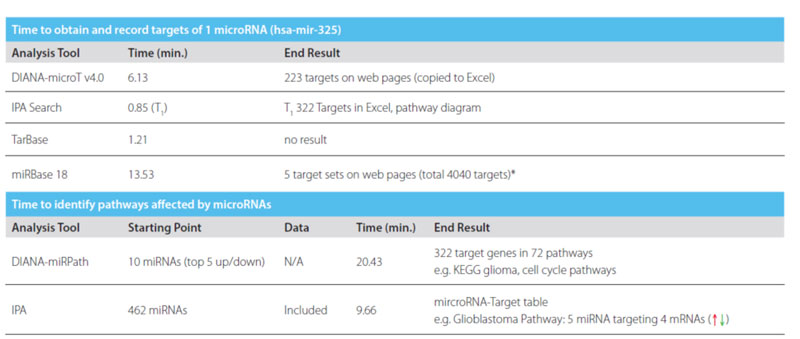
我们使用在一个在胶质瘤中表达最高的microRNA hsa-mir-325为例搜索。IPA只用了不到一分钟就得到了查询结果,而miRBase花了14分钟。IPA产生的结果来源于多种数据库,并且将靶向mRNA列表易于下载,并给出相应的文献支持。而miRBase却不能将搜索结果直接导出,只能手工摘取需要的信息(表二)。
不单单是搜索,我们也将胶质瘤的microRNA数据直接进行分析,希望能从中提取出来针对该疾病的有用的通路信息。DIANA-miRPath和IPA都预测了这些数据中会有的潜在的靶向基因和参与的通路。IPA花了大约10分钟完成了数据上传,靶向mRNA挖掘,并且直接提供了microRNA-mRNA之间的上调下调的预测结果。DIANA-miRPath不能直接上传数据,就只是人文的提供了一个microRNA的名单,因此,只有10个microRNA被分析了,并且耗时需要二十多分钟。其提供的结果是从不同的通路数据库中无筛选直接提取的。很难找出具有特异性的结果,增加了从中甄选有用信息的时间(表二)。
表一:显示查找并分析microRNA所需要的时间,计算了得到最初结果所需要的时间。
讨论:
本文的主要目的之一是将microarray或RNA-Seq实验的基因表达数据同时采用商业化和免费分析工具进行数据分析时所用时间进行定量比较。投资回报ROI值可以基于各个工具的分析时间计算得出。ROI由净收益除以软件花费计算得出。作为商业通路分析工具的先进供应商,IPA的用户数据分析平均量在2011年有20个左右。平均每个用户使用IPA进行数据分析的总时间数为62个小时。在本次测试中,IPA相对于其他软件综合起来(DAVID,PATHVISIO,Pubmed)大约每个课题节约了30个小时的研究时间(图四),相对于每人每个课题节约了约60%的分析时间。如果使用频率较高的话,相对于软件投资成本来说,节约的分析时间和提高的分析效率更加值得考虑。
另外,为了最大化高通量基因表达实验的价值,研究者必须将实验数据和生物学问题关联起来考虑。IPA帮助科学家们快速了解实验数据和生物学问题间的关系,并帮助他们快速产生有价值的推断和假设。我们总结了5个最有价值,最帮助IPA提高其ROI的因素。另外,IPA潜在价值在于能够帮助您更深入的了 解实验系统和模型背后的生物学意义,更好的评估实验结果的可信度,更好的帮助研究者筛选目标分子及做出判断,更能够在整个研究课题组中更好的分享结果,进行创新。
对数据分析重要的5个关键因素为:
1. 分析:阐释基因表达量发生变化的原因及其生物学效应;能够预测导致基因表达发生变化的上游调节子(转录因子、microRNA及其他分子)的活性状态是更好的理解生物系统和实验的影响的关键。
2. 分析:提供网络探索的系统生物学方法;采用迭代的探索方法帮助您 更深入的理解研究对象的生物学特性。 分析 工具同样需要具有多组学平台数据如转录组、蛋白质组、代谢组、microRNA等的联合分析能力。从而,科学家们可以使用工具进行相互作用网络生成,以及进一步的研究数据相关的生物学问题,如构建第二信使级联反应,鉴定临床可用的生物标志物或检测哪些信号通路发生了显著失调等。
3. 平台:支持专业的研究。在NGS技术等的推动下,相关的研究领域进展突飞猛进,良好的分析工具是能够紧紧关联专业的技术和知识,帮助分析复杂研究的数据。完整的分析工具应该可以同时帮助研究者分析RNA-Seq技术得到实验结果中的同源基因信息,并提供可视化的特定生物学功能与可变剪切及蛋白质功能域之间的相关性。另外,完整的分析工具应该也同时具有鉴定和筛选microRNA-mRNA配对情况,并筛选其中与目标通路和疾病相关联的具有生物学意义的结果
4. 整合:所有数据结果间的整合是非常耗时的。如果你在进行不同类型的分析时采用不同的软件,那样会花费您多的多的时间去进行数据整合和分析。而大多数软件往往也只具备某一类型的分析功能,而不是设计来回答您所有可能的问题。
5. 内容:需要完整全面、高时效性、高质量的数据库内容来辅助数据分析。对于分析软件来说,背景数据库的高时效性和内容高准确性是极其重要的。另外,背景知识是如何人工阅读提取和组织的同样对于数据库
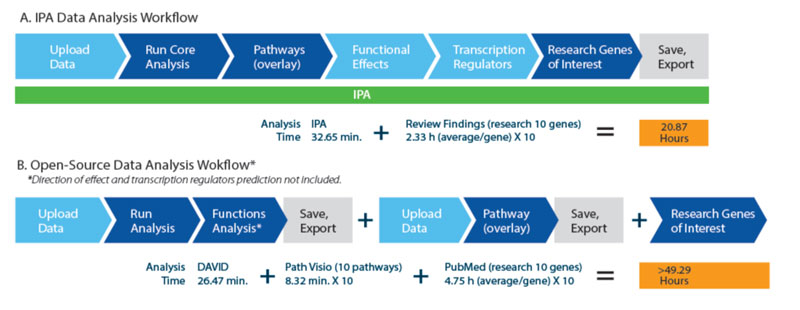
图四:IPA与免费数据分析软件的工作流程比较。A) 保守估计IPA数据分析加文献调研的时间约为20.87 小时,包括独有的生物学效应预测和转录因子调控网络预测B) 免费软件同样的工作流程需要用到3种不同的软件,包括功能和通路分析,通路查看和数据对接,人工文献调研等。该分析流程没有IPA所具有的上游转录因子调控状态预测,microRNA-mRNA功能及通路相关性筛选和下游生物学效应预测。
结论:
科学家们投入了大量的金钱和经历在高通量仪器设备上产出海量的数据,但是如果缺乏高效的数据分析手段的话,那么这些投资往往会高投入低产出。对于大数据平台如microarray或RNA-Seq,生物学解释工具如IPA是帮助科学家们快速筛选相关有意义信息和在统一的生物学背景中阐释数据意义的关键。在一个提供完整的上下游分子信息、分子相互作用网络,上游转录因子调控网络,下游生物学效应的软件环境中分析解释RNA-Seq或microarray数据集能够帮助您快速、可靠的找到复杂数据中的关键信息。采用如IPA这样的商业化软件可以使每个项目的分析时间节约超过30个小时,这样可以更快更好地优化后续的实验和建立假设模型。当您考虑选择一个更高回报率的分析策略是,最需要考虑的因素是如何最大化实验结果中的有价值信息,从而充分利用仪器设备和试剂的巨额投资。本文系统的描述和计算了microarray或RNA-Seq数据分析时三个关键步骤所需要耗费的时间及其投资回报,当使用IPA时,其分析效率和回报明显优于其它免费工具。另外,我们总结了五个在数据分析时需要关注的关键性因素,帮助您判断是否需要在数据分析时选择优质的工具。


