
Th3N4, Th2N3及Th2N2的第一性原理研究
1. 研究背景
近年来,钍基氮化物(Th-N)被广泛地应用于物理、化学和材料领域。钍基氮化物具有高熔点、高金属密度、高热导率、低蠕变速率以及良好的抗腐蚀能力的特点。因此,这类材料在核反应系统中,常被添加到锕系元素的混合亚临界氮化燃料里,用于核反应堆加速器的驱动。尽管目前有一些理论计算的工作研究了一氮化钍(Th-N)的性质,但是钍基氮化物家族的其他同晶型材料(Th2N2、Th2N3、Th3N4)依然缺乏相关的研究。因此,在本案例中,作者通过DFT方法,详细研究了这类Th2N2、Th2N3、Th3N4材料的几何结构性质、弹性性质和电子结构性质,并通过对价电荷密度的分析,深度剖析材料的成键方式。
2. 建模与计算方法
作者通过Welcome to MedeA Bundle中InformaticA数据库检索到了Th2N2(NH)、Th2N3、Th3N4的几何结构(见图1),并采用了GGA-PBE泛函,500 eV截断能来优化材料的结构,采用四面体计算方法来计算相关结构的电子态密度性质。

图1 (a)Th2N3(b)Th2N2(NH)(c)Th3N4的优化稳定结构。蓝色-Th;红色-N;白色-H
3. 结果与讨论
3.1 几何性质
作者通过MedeA-VASP模块做结构弛豫优化,并给出了结构参数信息(见表1)。
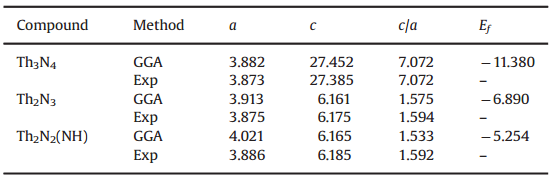
表1 Th-N系列化合物的计算晶格常数以及实验参数
在表1中,作者计算了晶格参数a、c、c/a的数值以及形成能Ef,可见,采用GGA的方法与实验值很接近。在Th2N2(NH)结构中,Th-N(1) 键长为2.922Å,Th-N(2) 键长为2.373Å,这一结果与XRD的测量结果几乎一致。
在表2中,作者计算了Th2N2、Th2N3、Th3N4结构中的原子位置,可以看到, Th3N4和Th2N2属于A-La2O3类型的晶体结构,分别在z方向存在2到3个内参数,而Th2N3则有2个内参数。
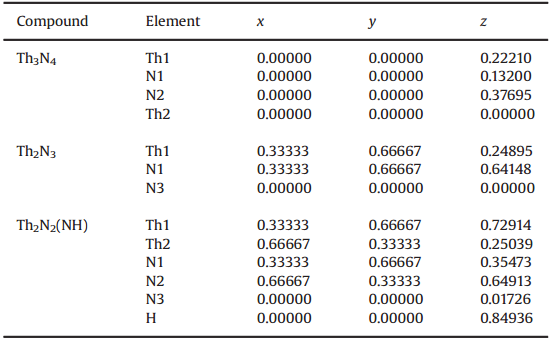
表2 Th-N化合物的原子晶格位置
3.2 力学性质
另外,作者通过MedeA-MT计算了Th-N钍基氮化物的弹性性质,由于同样计算了自选-极化效应(spin-polarization,SP)和自选轨道耦合(spin-orbit coupling,SOC)对材料的影响,发现其结果对几何结构,电子结构,弹性性质没有任何影响。因此,Th2N2、Th2N3、Th3N4被认为是非磁性体系(non-magnetic,NM)。在针对这3个Th-N钍基氮化物体系中,作者采用了0.05%应变系数,计算得到其弹性系数(Cij)见表3。
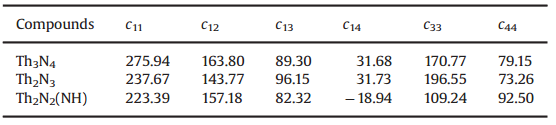
表3 Th2N2、Th2N3、Th3N4的弹性系数
Th-N钍基氮化物的力学性质结果同样列在表4中。通过与标准状态相比,形成这类化合物是能量稳定的。在体相中,金属Th和N原子的总能分别为 -7.443eV和 -8.316eV。于此同时,Th3N4具有最低的行程能。从德拜温度中也可以看到,Th3N4的温度最高为311.6 K, 说明该材料的硬度是最大的。
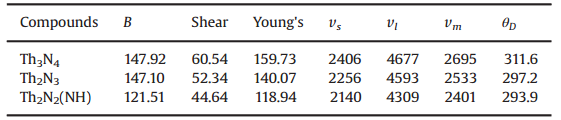
表4 Th2N2、Th2N3、Th3N4的体模量、剪切模量、杨氏模量的Hill数值(GPa),声速(m/s)以及德拜温度(K)
3.3 电子结构
作者同样用MedeA-VASP计算了Th-N钍基氮化物的态密度(DOS)。从图2(a)和(c)可以看到,Th3N4和Th2N2的禁带宽度分别为1.59 eV和 2.12 eV,其价带顶部主要由N-2p贡献,对于Th2N2其导带底部主要来自于Th的d轨道和f轨道的杂化和N-2p共同贡献,而对于Th3N4,其导带底部主要来自于Th的d轨道和f轨道的杂化,并且Th3N4的禁带宽度与光电子能谱得到的实验值1.7 eV非常吻合。除此之外,图2(b)显示的Th2N3的DOS跨越了fermi能级,说明了该材料具有一定的金属性,主要来自于N-2p态。Th2N2的绝缘性同Th2N3类似,这是由于Th2N2中的-NH基团平移价带导致的。值得关注的是,Th-N钍基氮化物的Th的p,d,f轨道都会与N-2p轨道产生一定的杂化。
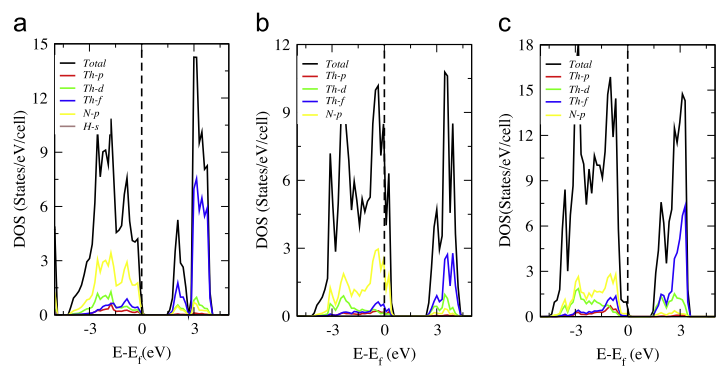
图2 (a) Th2N2 (b) Th2N3 (c) Th3N4的电子态密度。其中Fermi能级置于0 eV处
从化学的观点看来,不同体系的价带说明了物质的金属性还是绝缘性。然而,单纯看价带并不十分准确。Th3N4中,Th是+4价而N是-3价,Th有2个s电子,1个d电子,1个f电子,对于Th3N4化合物来说,其化合价是平衡的。N的成键态是完全的,因此Th3N4是有一定的间隙的材料。对于Th2N3来说,Th一共为8+(2×4+)而N为9-(3×3+),其电荷并不平衡,因此需要一个额外的H质子来平衡,这就产生了Th2N2(NH)这类结构。因此,在Th2N3结构中,Fermi能级就处在了N-p的带上,显示出了一定的金属性(见图2(b))。当引入一个H质子后,H-s态处于价带的上方,因此,又填充回了Th2N2的价带,此时,Th 的5f轨道又变成了空轨道,位于导带的位置。当然,其中包括了一些成键轨道和反键轨道,从图2(a)中可以显示出d和f的价带。
为了进一步说明成键的问题,作者又作了Th2N3、Th2N2、Th3N4的价电荷密度分布图,见图3,该Th2N3、Th2N2、Th3N4的价电荷密度分布图为沿着(110)平面的截面图,蓝色说明电荷密度减少而红色说明电荷密度增加。总体说来,N周围的电荷是增加的,Th周围的电荷是减少的,说明了电子有从Th往N迁移的过程,并说明了Th-N之间的化学键是离子键的成键方式。同样,从图3(b)中可以看出,由于有-NH基团的作用,电荷密度发生了一定的形变,说明该体系中,还存在一定的共价键的成分。
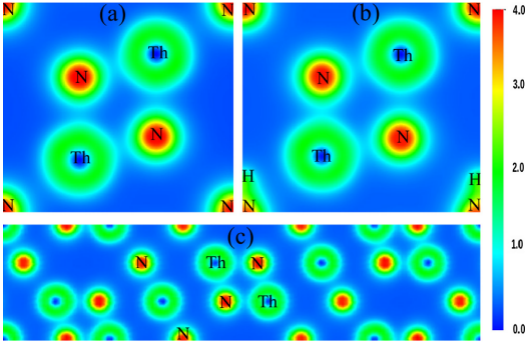
图3 (a) Th2N3 (b) Th2N2(NH)和 (c) Th3N4化合物沿着(110)面的价电荷密度分布
4. 总结与展望
本案例中,作者通过第一性原理DFT计算,详细地研究了Th-N钍基氮化物材料的几何结构性质、弹性性质和电子结构性质。Th3N4是最稳定一类氮化物,Th3N4和Th2N2是绝缘性的而Th2N3是金属性的,并理清了这一系列物质的电荷成键特性,这对于核反应堆的燃料材料的研究又是一个重要的贡献。
参考文献:
K.O. Obodo, N. Chetty. Ab initio studies of Th3N4, Th2N3 and Th2N2(NH). Solid State Communications 193 (2014) 41 –44.
使用MedeA模块:
Welcome to MedeA Bundle
MedeA-VASP
MedeA-MT


