
沥青质聚集行为(分子量及结构变化)敏感性的分子动力学研究
1. 研究背景
沥青质是重质原油中的主要成分,可溶于甲苯、烷烃溶剂及芳香烃类溶剂中。沥青质具有很大的分子量及复杂的聚合程度与结构,包含聚合芳香单体、饱和环链及烷烃支链等,主要元素有S、N、O。因此,在炼油过程中,沥青质的结构、聚合度、流体成分、外界条件(温度、压力、溶剂、工艺条件)等因素都会对油品的质量、稳定性等产生巨大的影响。 因此,为了更好的提升炼油工艺和工业效能,作者通过分子动力学模拟对沥青质聚集行为的敏感性变化做了十分有意义的研究。
2. 建模与计算方法
作者通过Welcome to MedeA Bundle中的Molecular Builder工具分别构建了沥青质的各个单体结构,包括有支链的烷烃链、羟基官能团、醚键结构、硫桥键以及硫醇基。
分子动力学的力场使用了MedeA-Force Field 中的PCFF+力场,PCFF+是基于全原子模型的动力学力场,能够很准确地预测流体的性质(密度以及内聚能)。PCFF+力场基于Lennard-Jones 9-6势函数,能准确的描述分子间以及分子内的色散-排斥作用。
2.1 分子动力学计算
作者采用了MedeA-LAMMPS进行分子动力学模拟,能准确描述短程相互作用,并用PPPM格点方法描述长程相互作用。比较重要的一点是,所采用的PCFF+力场是否能够准确预测烷烃和甲苯的输运性质,为此,作者在做LAMMPS模拟时,采用了 Green-Kubo方法进行平衡动力学计算,时间尺度上运行5 ns,并用MedeA-Viscosity预测沥青质聚合物的粘度。结果显示,预测的烷烃流体粘度能与碳原子数的变化关系趋势与实验值能够很好的匹配(见图1)。类似的,在250-400 K的温度范围内,采用PCFF+力场计算出的流体的剪切粘度的同温度的变化关系与实验结果也是完全一致的(见图 2)。考虑到溶剂的悬浊效应,初始优化的体系设置了较低的密度(0.08 – 0.15 g/ml)以避免分子之间的相互作用。
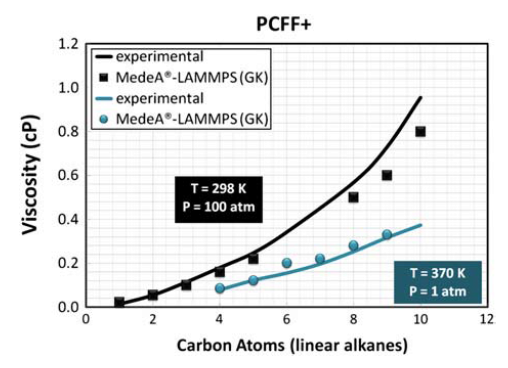
图1 采用PCFF+力场的烷烃剪切粘度随C原子个数的变化关系
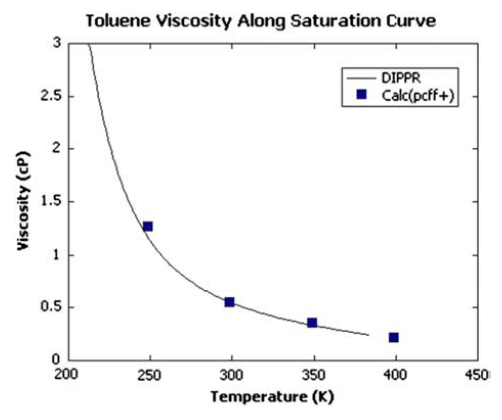
图 2 采用PCFF+力场以及平衡动力学Green Kubo计算得到的甲苯的剪切粘度随温度的变化关系。
每一步的分子动力学计算流程如下:
在常数体系下进行能量最小化;
设置外界温度为350 K,并根据温度调节选择初始速率,在这个计算过程中,无初始速率;
在每一个动力学模拟运算阶段设置NPT系综,压力的变化从 200 bar 下降到 1 bar, 运动时间为 100 ps,并采用Nose Hoover 恒温模型调温,初始采用 200 bar的外压是为了加速在模拟适当密度情况下的收敛;
最后MD的计算结果由MedeA-Environment 来可视化。
图 3 (a)展示了沥青质分子的聚合物结构。图 3(b)展示了纳米聚合物沥青质层最小间距的不同尺寸分布。 事实上,堆叠层的芳香烃单元结构对于沥青质的聚集有很大的影响。其中,最小的堆叠层间距仅有0.4 nm,略大于石墨烯层的间距,因为在沥青质中,含有S、O、N等元素以及饱和的碳环,会有较强的相互作用。
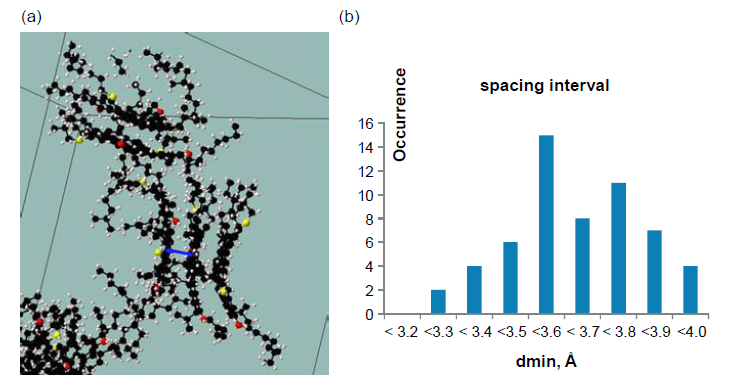
图3 (a)3个沥青质分子聚集的溶剂模拟结构,每片薄层结构两者间由C原子连接呈现堆砌结构。(b)每层沥青质层的最小间距的分布比例
2.2 分子模型
沥青质是重质原油的主要成分,其中,各类元素的比例为H/C=1.2, S/C= 0.02, O/C= 0.02, N/C= 0.01。表1 和图4 展示了沥青质不同结构的分子量和结构模型。
表1 沥青质不同结构模型的分子量和元素比例
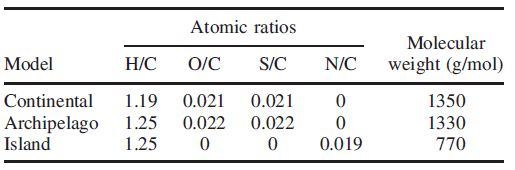
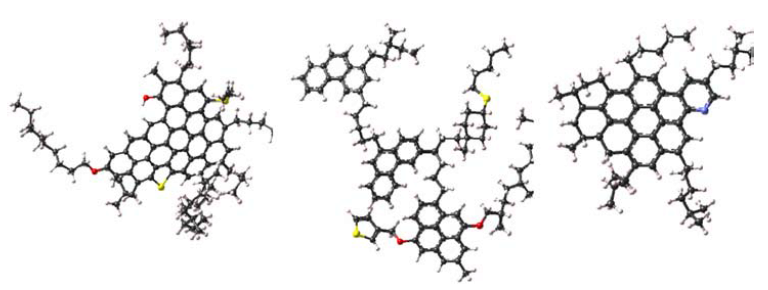
图 4 沥青质分子结构模型: (左边)continental modl大陆式模型(一部分平面,一部分堆砌);(中间)archipelago model分散模型(比较平缓、分散);(右边)island model小岛模型。图中:黑色-C;黄色-S;红色-O;紫色-N。
总体看来,沥青质分子包含了稠环芳烃的骨架以及烷烃链结构。较大尺寸的模型意味着烷烃链中的C原子个数较多(包括支链)。当不同的分子构型自聚集或者共混聚合的时候,会表现出不同的理化性质。
2.3 模拟条件以及初始化结构
本案例中,主要涉及了两种溶剂: 甲苯和正庚烷。温度条件为 350 K,压力为1 bar。模型结构为600个分子(甲苯和正庚烷)作为溶剂与9个沥青质分子共混。整个slab的大小为10 nm × 10 nm × 10 nm。 在NPT模拟时,密度增加,slab的体积变为5 nm × 5 nm × 5 nm。同时,考虑到初始构型的影响,9个沥青质分子(大陆式模型)的构型为处于平行于Z轴方向的一列结构,厚度为13 nm,周围由648个甲苯分子包围,组成 13 nm × 13 nm × 13 nm的盒子大小。这样就避免了聚合物聚集带来的影响。
3 计算结果与讨论
在每一种溶质沥青质分子与溶剂的系统中,其内能与密度在模拟了> 10 ps 后都能到达一个稳态。随后在一定的幅度内波动。再之后的 1 ns 时间内,便不会出现很大的密度变化了。经MD 模拟的结果可知,甲苯溶剂体系的平均密度(0.82 -0.84 g/ml)要明显高于正庚烷溶剂体系的平均密度(0.66 -0.69 g/ml)。这或许是由不同的溶剂分子的极性所影响的。
图5和图6分别显示了沥青质分子在正庚烷溶剂和甲烷溶剂中经过动力学模拟后各个构型的模型图。在分散模型中,局域的芳香烃分子团簇是由不同的分子单元构成的。
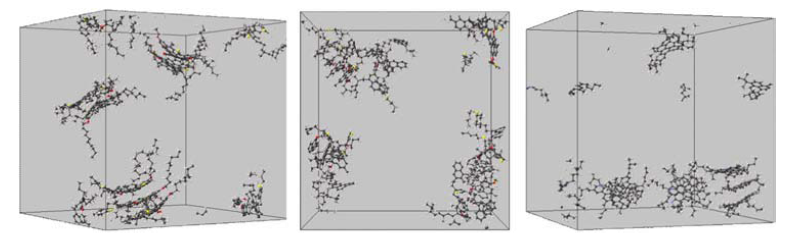
图 5 9个沥青质溶质分子于600个正庚烷溶剂中在350 K,1bar, NPT模拟15 ns后的结构模型。(溶剂正庚烷分子未显示)。左图:大陆式模型,显示了4个、3个、2个分子堆积的沥青质模型;中图:分散模型,显示出了若干小原子簇堆积模型;右图:小岛模型,有2至3个沥青分子堆叠成团簇。
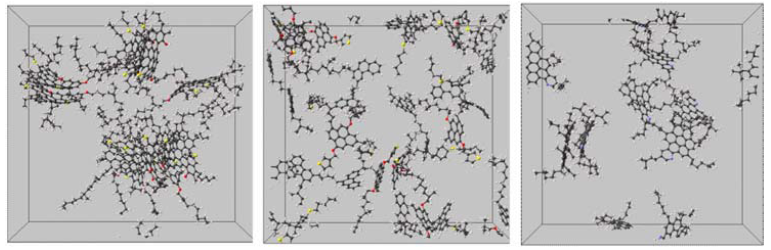
图 6 9个沥青质溶质分子于648个甲苯溶剂中在350 K,1bar, NPT模拟15 ns后的结构模型。(溶剂甲苯分子未显示)。左图:大陆式模型;中图:分散模型;右图:小岛模型
同时,作者又增加对构型初始化,在做动力学模拟的测试。初始的分子队列构型会在逐渐模拟的过程中渐渐消失(见图7),说明模型是否初始化对整个动力学过程的影响非常小。因此,该模拟过程说明了包含了4个分子以上的纳米聚集并不是十分稳定的,从另一个角度讲,各个分子簇的聚集会受到一个能垒的阻碍。

图7 9个沥青质溶质分子的初始化构型影响(大陆式模型)。溶剂为648个甲苯分子,350 K, 1 bar。(溶剂分子甲苯未显示)左图:100 ps 动力学模拟后的沥青质分子堆积情况;右图:动力学模拟了 1 ns后的构型,显示了3个完全不同的团簇。
不考虑溶剂的因素,作者又考察了溶质分子团簇的粒径同动力学模拟时长的关系(见图8)。图8说明了沥青质溶质分子和溶剂分子相互聚集而产生的不同特性。大陆式结构模型的分子团簇粒径会随着模拟时间的增加而持续增大。在正庚烷溶剂中,最后的跳跃点来自于两个堆积团簇在运动了14 ns至 15 ns以后聚集成了四堆积的团簇。然而,这仅仅是单个分子团簇的观测到的情况,同样的高聚集现象并没有在沥青质分散型结构和小岛型结构中出现。
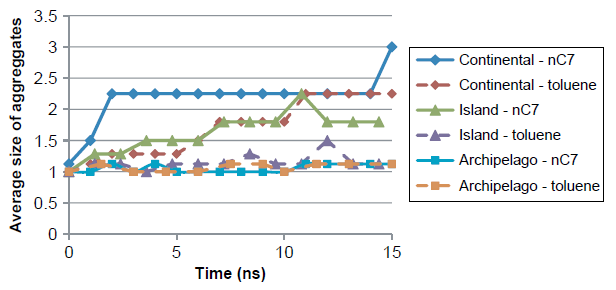
图 8 三种沥青质分子聚集的的平均团簇尺寸随着模拟时间的变化关系。体系为:9个沥青质分子在600个溶剂分子里,模拟条件为350 K,1 bar
另外,作者发现,小岛模型结构在正庚烷溶剂中的聚集作用是非常微弱的而在甲苯溶剂中的聚集效果却很强(见图9)。
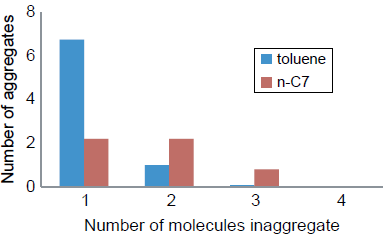
图 9 小岛式模型沥青质分子分别在甲苯和正庚烷溶剂中的粒径聚集尺寸分布
沥青质分子在各类溶剂中的扩散性质也是非常重要的,因此作者又通过MedeA-Diffusion模块计算了沥青质分子在正庚烷中的扩散系数(见图10)。从图中可知MSD与模拟时间有很好的线性关系,意味着沥青质遵循了沿着各个方向的各向异性的Fickian扩散规律。根据Einstein方程计算得到扩散系数为 4.32 × 10 -5 cm2/s。
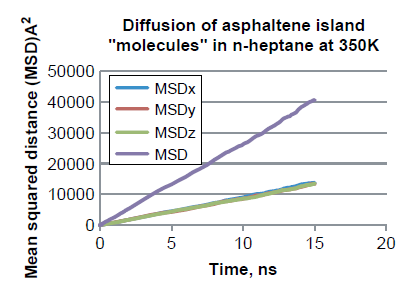
图 10 芳香烃单体中C原子的均方根位移(MSD)随着时间的变化关系(溶剂为正庚烷)
4. 总结与展望
本案例中,作者重点研究了重质原油主要成分沥青质分子的聚集方式和结构行为。不同的分子构型会产生不同的聚集表现行为,从聚集粒径分布,粘度性质和扩散性质等各方面来看,小岛模型的沥青质分子在正庚烷溶剂中的聚集效果是理想的。通过对沥青质分子的聚集方式的研究,能更好的有助于对石油化工中炼油工艺的改进。
参考文献:
Philippe Ungerer, David Rigby, Benoit Leblanc, Marianna Yiannourakou. Sensitivity of the aggregation behaviour of asphaltenes to molecular weight and structure using molecular dynamics. Molecular Simulation. 2014, 40(1-3), 115-122.
使用MedeA模块:
Welcome to MedeA Bundle
MedeA-Amorphous Builder
MedeA-LAMMPS
MedeA-Forcefield
MedeA-Viscosity
MedeA-Diffusion


