
基于高熵催化剂的电池驱动N2电解:从理论预测到原型模型
关键词:高熵材料,电催化,氮还原反应(NRR),密度泛函(DFT)
1.案例背景
氨 (NH3) 是生产肥料和其他工业化学品至关重要的材料。目前可以通过 Haber-Bosch 工艺在高温 (350-550 ℃) 和压力 (150-350 atm)下生产NH3,需要基础设施极为严苛。这意味着 NH3 通常是在大型集中设施中批量制造。相比之下,如果 NH3 可以在要求较低的条件下生产,使用较小的设备去生产少量 NH3 供单个地方使用,这可以降低大规模存储、运输和相关过程的大量成本。研究表明,电化学是实现此目的的一项先进技术,它具有相对简单地反应装置和易得的原材料。以水 (H2O) 和氮气(N2) 作为反应物,在电解槽中以催化剂促进N2电解,从而环保的生产 NH3(N2 + 3H2O → 2NH3 + 3/2O2)。高熵 (HE) 材料,尤其是高熵氧化物 (HEO),代表了一类由超过五种金属或金属阳离子组成的材料,由于几乎无限可能的元素组合,因此可以应用于各种领域(例如催化、能量存储和热电)。本案例中,作者利用DFT计算预测了高熵氧化物 (HEO) 能够有效促进 N2还原反应(NRR) 和 析氧反应(OER) 。随后通过实验合成了HEO,其形态为由超薄纳米片组装而成的海胆形中空纳米球,且具有出色的电催化活性。而后进一步使用DFT计算了HEO的NRR反应的路径的吉布斯自由能,解释了其高效NRR的原因。
2.建模与计算方法
作者通过MedeA Environment 创建了由1/3的M(II)O4和2/3的M(III)O6组成的单相尖晶石结构形式的HEOs理论模型,不同金属间通过共享O原子相连( M = Ni、Co、Fe、Mn、V,Ni:Co:Fe:Mn:V 的原子比为 1:1:1:1:1);随后采用MedeA-VASP模块中的DFT方法对HEOs结构进行优化,截断能选取400 eV,K点基矢选取3×2×1;同时计算HEOs的电子结构和NRR反应路径的吉布斯自由能,揭示了其高效催化反应的原因。
3.结果与讨论
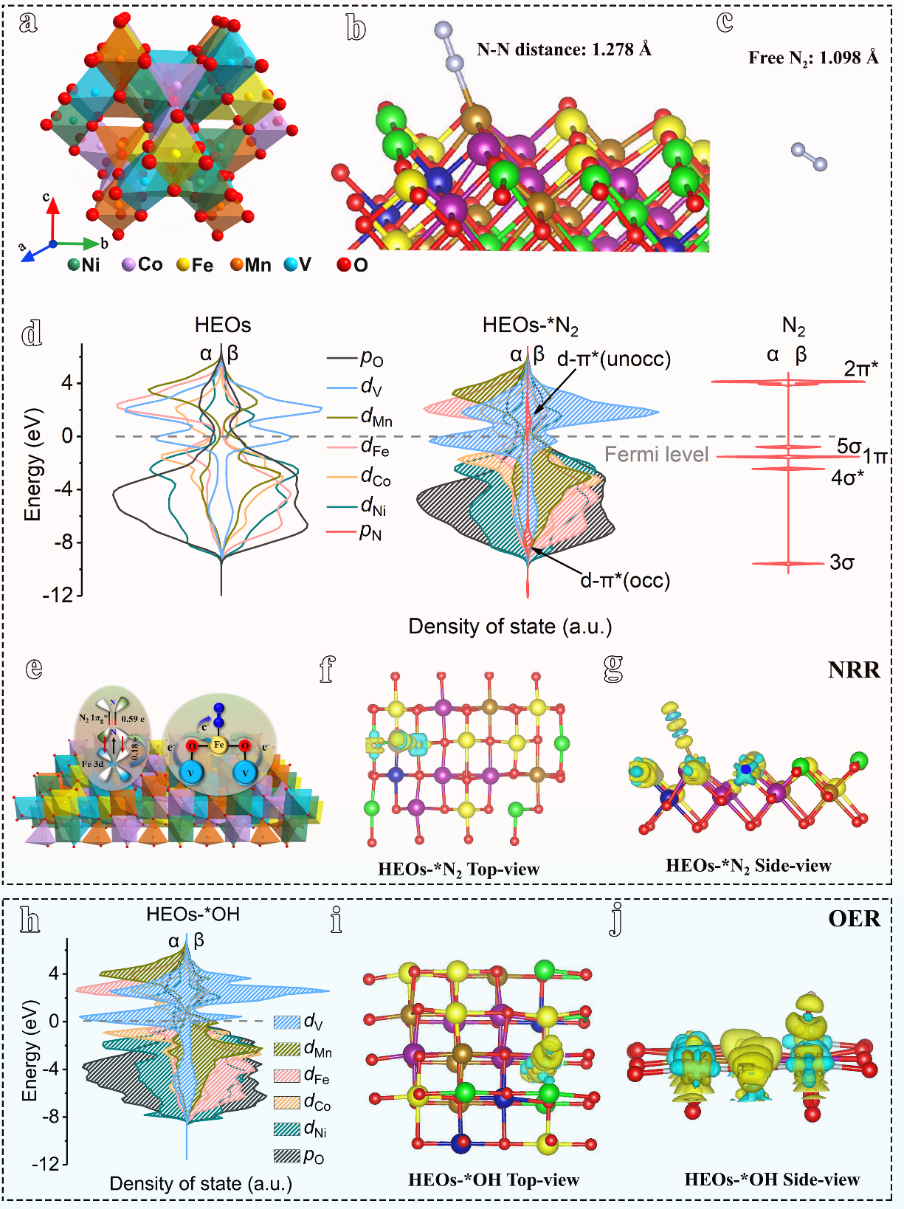
图1 HEO 上阴极 NRR 和阳极 OER 活性的预测。 (a-g) 阴极 NRR 活性的预测。 HEOs(a)、*N2(b)、游离N2(c)的晶体结构模型; (d) N2 吸附前后HEO费米能级附近的态密度 (DOS),以及与游离 N2 进行比较; (e) N2 吸附 HEO 的简化示意图; *N2的差分电荷密度图:顶视图(f)和侧视图(g); (h-j) 阳极 OER 活性的预测:(h) OH-吸附后 HEO 费米能级附近的 DOS;*OH差分电荷密度图:顶视图 (i) 和侧视图 (j)。黄色和青色区域分别代表电荷积累和耗尽。
对于 NRR 过程,N2 分子的活化是非常具有挑战性的,这是后续氢化步骤的先决条件(图 1b)。 因此,作者使用MedeA-VASP计算了HEO材料吸附N2前后的电子性质。首先是通过态密度(DOS)和差分电荷密度来预测N2 在 HEO 表面的活化能力。DOS显示HEO费米能级附近的顶价带主要由 Ni d、Co d、Fe d、Mn d、V d 和 O p 价轨道组成,且Fe 是 NRR 的活性中心(图 1d)。 这种高密度的价电子轨道表明 HEO 具有高电子转移能力,这可以促进 NRR 反应的 N2 活化(图 1d)。在HEO吸附N2后,N2 p 轨道的能级与 Fe d 轨道重叠,这导致 d-π* 轨道被部分占据(图 1d)。特别是,在 N2 吸附后,V d 的价电子 α 和 β 轨道移动到费米能级附近且高自旋极化,表明弱化 N≡N 三键对 NRR 具有显着的协同效应(图 1d)。因此,N≡N 键在吸附后从 1.098 拉长到 1.278 Å(图 1b、c )。Bader显示HEO上0.59 |e|电子转移到N2上,这是因为 Fe 3d 轨道上的电子被捐赠给 *N2 的反键轨道 (π*)(图 1e-g)。基于上述分析,我们预测 HEO 可以为 NRR 活化N2。随后作者在实验上合成了 HEO,其形态为由超薄纳米片组装而成的海胆形中空纳米球。经检测, HEO 对 NRR(NH3 产率:47.58 μg h-1 mg-1 和法拉第效率(FE):10.74%)和 OER(226 mV @10 mA cm-2)的出色电催化活性。
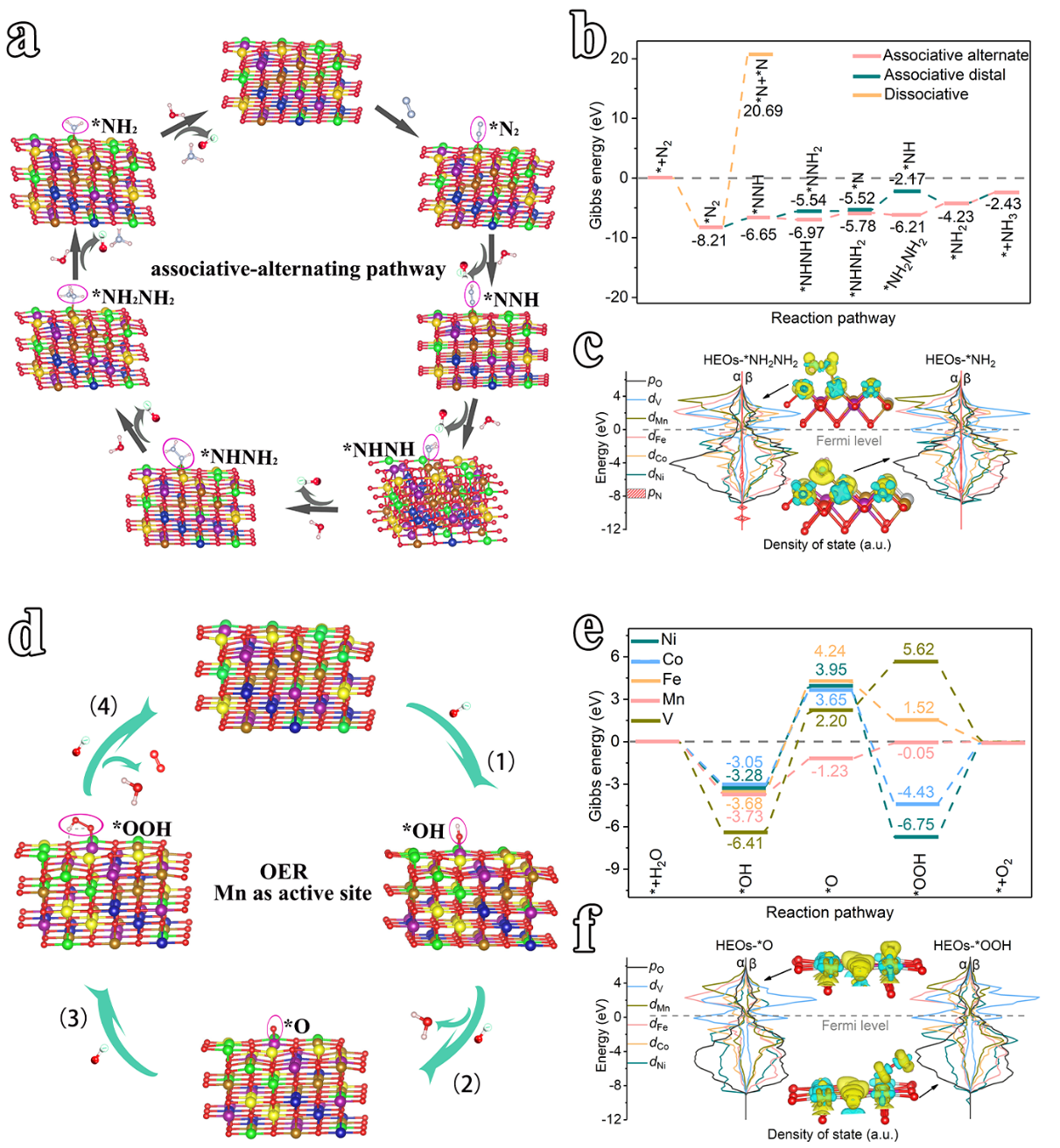
图2 HEO上NRR 和 OER 的 DFT 计算。 (a) NRR 的关联交替反应路径; (b) NRR 自由能图; (c)从*NH2NH2到*NH2的NRR电位决定步骤的态密度(DOS)和差分电荷密度图; (d) OER 的反应路径; (e) 不同金属位点的 OER 自由能图; (f) OER 电位决定步骤从 *O 到 *OOH 的 DOS 和差分电荷密度。
为了进一步了解HEO催化剂上NRR 和 OER 的反应机理,使用MedeA-VASP计算了两反应路径的吉布斯自由能。首先,研究了NRR路径(即分离、远端缔合和交替缔合)(图 2a-c)。如图 2b 所示,HEO 可以自发吸附N2分子,其 ΔG为-8.21 eV,这证明N2被轻松激活。接下来,对于靠前加氢步骤(*N2 + H2O + e- → *NNH + OH-),缔合路径的能垒(1.56 eV)远低于解离路径的能垒(28.90 eV),表明缔合路径为本步反应步骤(图 2a、b)。此外,对于第二个氢化步骤,交替缔合路径(*NNH + H2O + e -→ *NHNH + OH-,-0.32 eV)中所需的能垒小于远端缔合路径(*NNH + H2O + e -→ *NNH2 + OH-; 1.11 eV),表明交替缔合路径更适合后续的氢化反应。随后研究了HEO上NRR 的关联交替路径。 *NHNH 中间体捕获第三个 (H2O + e-) 对形成*NHNH2 ΔG为1.19 eV,然后 *NHNH2 中间体的捕获第四个 (H2O + e-) 对以形成*NH2NH2 ΔG为-0.43 eV,表明该氢化步骤是放热的并且自发发生(图2a、b)。 此外,*NH2NH2中间体通过连续吸附两个(H2O+e-)对进行氢化,生成NH3分子,生成的两个NH3都具有高能垒(1.98和1.80eV)。这证明了电位决定步骤(PDS)是*NH2NH2的氢化( *NH2NH2 + H2O + e- → *NH2 + NH3) 而不是文献中的N≡N 裂解。
为了解释HEO上多金属位点之间协同效应的增强 NRR 催化活性,计算了 DOS 和差分电荷密度( *NH2NH2 和 *NH2,图 6c)。首先,*NH2NH2 α和β-spin π轨道的能级显示与HEO的所有金属d轨道部分重叠,表明HEO的多金属位点可以优化氢化步骤,通过协同调节行为促进分子NH3的吸附解吸和电子转移(图 6c)。同样,差分电荷密度图图也显示 N 原子、Fe 和相邻金属原子失电子,表明 Ni、Co、Fe、Mn 和 V d 轨道之间的多金属协同效应可以促进电荷的转移,从而有利于加氢步骤。此外,对于*NH2中间体,*NH2的α和β-自旋π轨道与HEO的金属原子轨道大量重叠,这能够进一步促进Fe位点与吸附的N之间的相互作用和电子转移,从而促进了后续的氢化步骤,增强NRR活性。
4.总结与展望
本案例中作者对 HEO 的理论预测、实验分析和应用提出了系统研究。 从理论上讲,发现 HEO 可以同时激活 N2 以实现 NRR,并通过高 DOS 和独特的 α 和 β 轨道自旋极化过程加速 OER 的羟基去质子化。 然后,通过一种简单方法合成了HEO,其形态为由超薄纳米片组装而成的海胆形中空纳米球。由于独特的电子结构, HEO 对 NRR、OER 和 N2 电解表现出优异的催化活性,具有显着的 NH3、FEs 和 EEs产率。本工作可以为广泛的实际应用的催化剂设计和装置制造提供新的线索。
参考文献:
Y. Sun, L. Yu, et. al,Battery-Driven N2 Electrolysis Enabled by High-Entropy Catalysts: From Theoretical Prediction to Prototype Model, Small, 2022, 18:2106358
使用MedeA模块:
MedeA Environment
MedeA VASP


