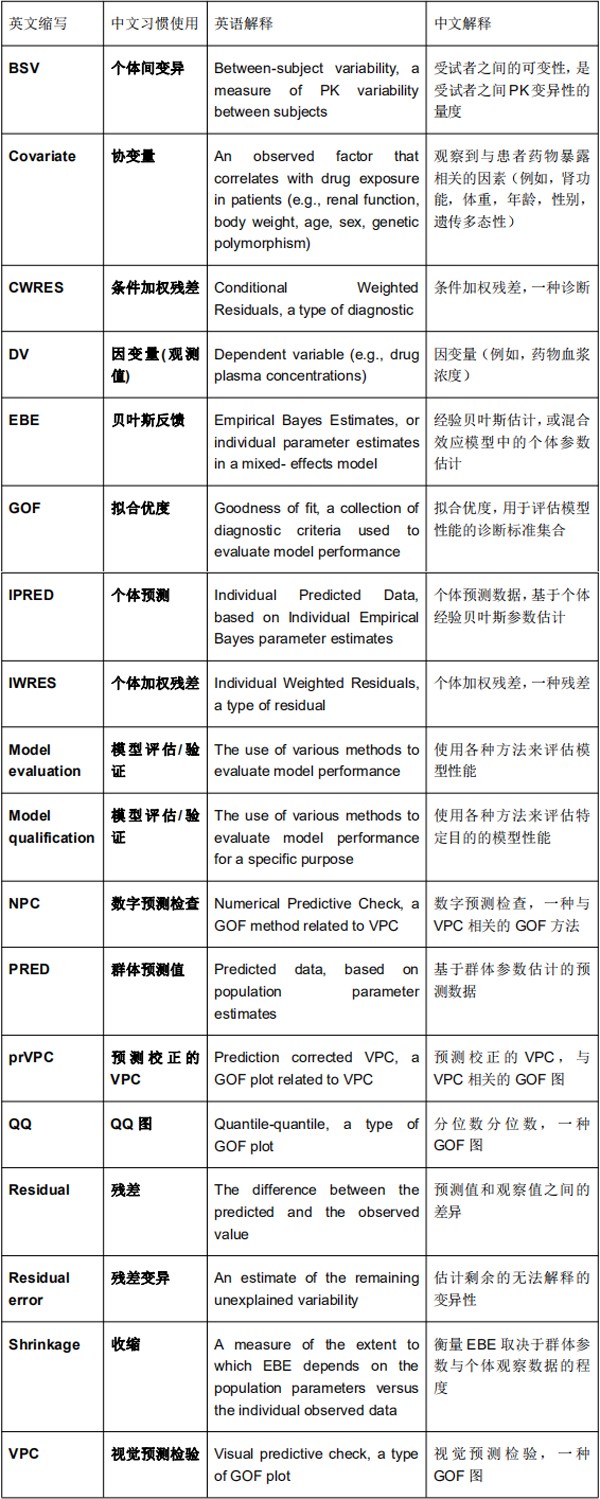
群体药物代谢动力学行业指南
该指南草案最终定稿后,将代表美国食品药品管理局(FDA或FDA)对该主题的当前想法。它不为任何人建立任何权利,也不对FDA或公众具有约束力。如果满足适用法规和规定的要求,您可以使用替代方法。要讨论替代方法,请联系标题页上列出的负责本指南的FDA工作人员。 Ⅰ. 引言
本指南旨在帮助新药申请(NDAs)和生物制剂许可申请(BLA)的申办方应用群体药代动力学(群体PK)分析。群体PK分析经常用于指导药物2的开发并提供关于个体化治疗的建议(例如,通过定制的剂量)(Marshall等人2015; Lee等人2011; Bhattaram等人2005)。在上市申请提交中,对群体PK数据充足的收集和分析,减轻了对上市后需求(PMR)或上市后承诺(PMC)的需要。
该指南包括群体PK分析的常见应用,为药物开发和药物使用提供信息。该应用列表并不是全面的,而是提供说明性示例。该指南还包括FDA目前对支持监管决策所需的数据和模型要求的思考,和对申办方基于群体PK分析的药物说明书的建议,以及对提交给监管机构的群体PK报告的格式和内容的一般期望。
一般而言,FDA的指导文件不构成法律上可执行的责任。相反,指南描述了监管机构目前对某一主题的看法,应仅视为建议,除非引用了具体的法规或法定要求。在机构指南中使用“应该”一词意味着建议或推荐某些内容,但不是必需的。
注释
1.本指南由美国食品药品管理局药物评价中心(CDER)和研究中心的临床药理学办公室和生物制品评估与研究中心(CBER)编写。
2.就本指南而言,对药物,药物和生物制品的引用包括根据“联邦食品,药品和化妆品法案”(FD&C法案或法案)第505条(21 USC 355)批准的药物和根据351的作为药品的公共卫生服务法(PHSA)(42 USC 262)。
Ⅱ. 背景
群体PK分析是一种成熟的定量方法,可以量化和解释个体间药物浓度的变化(Sheiner,Rosenberg和Marathe,1977; Grasela Jr和Sheiner,1991)。遵循相同给药方案的个体之间的药物浓度可以显着不同。变异性可归因于内在的患者因素,例如肝脏或肾脏损伤的存在和程度或遗传多态性的存在,或外源性患者因素,例如食物消耗或可能与施用的药物相互作用的伴随药物。在某些情况下,内在或外在因素导致药物浓度具有临床意义上相关的改变,这需要临床管理策略,例如剂量或给药方案的变化。
在独立的临床药理学研究中经常会研究常见的影响药物暴露的内在和外在因素。3 独立研究得到很好的控制,可以对这些相互作用进行有力的评估。然而,独立研究通常旨在关注具有最大潜在影响药物暴露的内在和外在因素,使许多可能的相互作用未被研究。群体PK分析通常包括直接从患者收集的数据,允许评估在健康志愿者中未另外评估的多种内在和外在因素。此外,包含在群体PK分析中的相对大量的患者,可以提高影响药物暴露的因素的估计效果的准确度,并确认哪些因素不会改变药物暴露。
注释3,见IX。针对独立临床药理学研究的行业特定FDA指南的参考文献。
群体PK分析整合了一系列剂量的所有相关PK信息,以确定可能影响药物暴露的因素。此类信息可来自单剂量或稳态后,密集的PK采样或稀释的PK采样,以及健康个体或患者群体的研究。反过来,这些分析可以为这些场景提供信息:为特定亚群的定制给药策略,规划后续研究或药品说明书撰写。
Ⅲ. 群体PK分析的应用
鼓励申办方在与监管机构的适当里程碑会议上,寻求关于使用群体PK分析进行药物开发决策或回答监管问题建议。申办方应联系临床药理学办公室,讨论群体PK分析的新方法和应用,以便为药物开发和使用提供信息。
对特定群体进行PK分析以支持预期目标的信心增加如下:
了解药物的PK特性
研究方案或数据分析计划中的预先指定的问题将通过群体PK分析来解决
足够数量和质量的PK数据,代表指定的群体和相关的感兴趣的子群体
良好的模型性能(即,模型应该以可接受的偏差和精度描述数据)并且对于预期目的是有效的
A. 群体PK分析在药物开发中的应用
1.选择在临床试验中进行测试的剂量方案
群体PK分析可以识别显着影响PK变异性的协变量,并通知给药方案在临床试验中进行测试,以帮助最小化患者的治疗反应的变异性。例如,观察到的体重和药物暴露之间的强关系可以为基于体重的给药方案提供支持(例如,基于体重切点(body weight cutpoints)的mg / kg给药或分类给药)。此类分析应结合对药物暴露与药物作用(例如,通过使用药效学生物标记物或临床终点),靶标参与(例如,受体占据)或药物毒性之间的关系的充分理解,以通知并进一步改进剂量。
群体PK模型还可用于模拟在先前临床研究中未直接研究的剂量或给药方案后预期发生的药物暴露(关于模拟策略的讨论,参见VD部分)。例如,群体PK分析可用于预测由于包含负荷剂量,改变剂量或改变给药方案的给药频率而导致的PK变化,以用于药物开发计划中的后续试验。在极少数情况下并且在适当合理的情况下,此类分析与暴露 - 反应数据一起用于批准尚未在临床试验中直接评估的给药方案(Kimko和Peck,2010)。建议申办方为此类申请寻求机构的意见。
2.推导样本量和抽样方案要求,以促进协变量效应的可靠估计
使用群体PK模型进行的模拟可以帮助确定在给定定义的协变量效应大小的情况下获得足够的统计效力来检测显着的协变量所需的亚群中的患者数量(例如,接受需要包括的伴随药物的患者的数量)检测显着的药物 - 药物相互作用的分析;见第III.B.2节)。模拟和优化设计方法可以最大化这种分析的效用。例如,可以优化研究样本大小和PK采样时间表,以便可以以确定的准确度估计PK参数(有关各种采样时间表的讨论,请参见第IV.B部分)。
3.推导暴露指标进行暴露 - 反应分析
2003年FDA题为“ 暴露 - 反应关系 - 研究设计,数据分析和监管应用”的行业指南概述了新药暴露 - 反应(ER)关系的重要性和应用。4 群体PK分析可用于推导可用于进行连续ER分析的患者PK暴露指标。
备注4,我们会定期更新指南。有关指南的最新版本,请访问FDA指导网页:https://www.fda.gov/RegulatoryInformation/Guidances/default.htm。
推导的暴露度量(例如,曲线下面积(AUC),最小药物浓度(C min ))可以用作稳态患者的平均药物暴露的量度。暴露指标的推导应考虑到:(1)剂量中断或修改; (2)药物的药代动力学随时间,疾病状态或疾病严重程度的变化。
群体PK模型可以预测特定时间点的个体患者暴露,而不管采样时间的扩散(例如,可以预测所有受试者的谷浓度)。
当PK数据在少数受试者中缺失时,群体PK模型可以基于受试者的个体协变量(例如,体重,遗传多态性,性别)预测最可能的浓度 - 时间曲线。假设残余误差和受试者之间的变异性较低,并且观察到的协变量对药物PK性质的影响很大,这种预测是有用的(参见VD部分)。
基于经验贝叶斯估计(EBE)生成个体PK患者暴露度量。当个体数据稀疏或无信息且参数收缩率很高(即通常大于20%至30%)时,EBE被认为不太可靠(Savic和Karlsson 2009)。除参数收缩外,个体PK患者暴露指标的可靠性取决于收集的PK数据的性质和模型假设的有效性(例如,时间不变的药代动力学,模型结构,剂量比例药代动力学)。有关模型验证的讨论,请参阅VC部分;有关用于群体PK分析的数据充分性的讨论,请参见第IV部分。
4.儿科研究设计
2014年FDA工业指南草案中概述了使用建模和模拟来为研究设计提供信息并优化儿科患者的剂量选择,该指南题为“ 药物和生物制品儿科研究的一般临床药理学考虑因素”。5 文献中提出了儿科研究的其他样本量考虑因素(Wang等,2012)。群体PK分析在儿童中尤其适用,因为与传统PK分析相关的丰富采样相比,它允许使用不频繁(即稀疏)的采样,从而最小化采样的血液总量。由于从儿科患者获得的血液样本数量有限,通常预计儿科研究中的取样窗口比成人研究中的取样窗口更宽。
备注5,最终,本指南将代表FDA目前对该主题的看法。
选择用于儿科研究的给药方案可以通过使用利用成人PK数据开发的群体PK模型进行模拟并且结合:(1)异速生长的原理; (2)可影响药物药代动力学(个体发育)的发育变化的知识; (3)儿科制剂的生物利用度数据(Holford,Heo和Anderson 2013; Barbour,Fossler和Barrett,2014; Zhang等,2015; Mahmood,2014)。
包括群体PK模型中对不同年龄,特别是2岁以下儿童患者生理成熟的最新理解,可进一步提高识别适当儿科剂量的能力。应该注意的是,剂量选择还需要了解成人和儿科的疾病相似性和ER关系(参见2014年FDA工业指南草案,题为“ 药物和生物制品儿科研究的一般临床药理学考虑因素” 6)。
备注6,最终,本指本指南将代表FDA目前对该主题的看法。
B.群体PK分析在药物使用中的应用
使用来自晚期临床试验的数据的群体PK分析以及独立临床药理学研究的结果可用于支持药物说明书中的吸收,分布,代谢和排泄(ADME)信息。在某些情况下,使用非房室模型分析(NCA)方法的传统PK数据分析是不够的。例如,这样的研究设计可能是非常困难,为具有长半衰期的药物设计研究,以允许的由AUC 0-t(从零到一个确定的时间点的浓度-时间曲线下面积)外推,估计得的AUC外推面积小于AUCinf(以小于从零时刻至无穷大时刻的浓度-时间曲线下面积)面积的20%。应使用群体PK方法分析此类研究(Svensson等,2016)。
此外,常规使用群体PK分析以及来自独立研究,巢式研究或其他来源的相关信息来评估协变量对药物及其相关代谢物的PK参数的影响以支持给药建议。由于协变量对药物暴露的影响,需要进行剂量调整,这应该在已知的ER关系的有效性和安全性的背景下进行解释。ER关系应用于确定剂量调整与临床无关的浓度边界(即,这些边界内的协变量效应可能无法保证剂量改变;但是,这些边界外的协变量效应可能需要剂量调整以优化益处 - 药物的风险特征)。
1.特定群体
可以将来自群体PK分析的结果纳入药物产品说明书中以描述一般患者群体或特定群体中的PK特性。基于群体PK分析的结果,对特定群体进行额外的标注说明,通常包括描述协变量效应大小的语言(language),评估具有临床意义上相关的改变,并且可以包括关于剂量调整的需要或缺乏的建议(参见第六节了解更多信息)。协变量分析是否支持在说明书使用额外的语句说明取决于多种因素,包括分析中包含协变量的受试者数量,下面列出了这些情况的一些示例:
- 某些药物(例如,剧毒剂)在单独的肾脏或肝脏损伤,没有感兴趣的医学病症的患者中,进行研究可能与不符合道德标准。在这种情况下,在具有内在因素的临床试验中充分代表患者,以及足够的PK取样以可靠地表征内在因素效应,可以允许使用群体PK分析来为这些患者提供药品说明书。
- 在特定群体(例如,患有不同程度肾损伤的患者)可以安全地纳入晚期临床试验的情况下,群体PK分析可用于表征药物的暴露及其与反应的关系,并在该群体中得出剂量推荐。
- 传统上,在独立的临床药理学研究中没有研究一些确定的特定群体,因为缺乏对药物药代动力学有很大影响的先验假设。相反,通常在没有独立试验的情况下研究诸如性别,年龄,体重或种族对研究药物的药代动力学的影响等因素。可以想象,群体PK分析可用于描述这些亚组中药物的药代动力学。
- 从儿科和成人数据建立的群体PK模型的模拟可用于比较药物在儿科患者和成人中的暴露,以得出推荐的儿科剂量用于标记。
2.药物-药物相互作用
可以使用群体PK分析来评估临床DDI(例如,作为阶段3研究的一部分的嵌套研究(nested studies))。嵌套DDI研究的一般设计考虑因素可在2017年FDA工业指南草案中找到,该指南题为“临床药物相互作用研究-研究设计,数据分析,剂量影响和药品说明书建议”。7 使用群体PK方法来表征药物的DDI潜力并非没有限制,并且在其他地方已经描述了使用群体PK分析来评估DDI的方法学考虑(Bonate等人2016; Wang等人2017)。使用群体PK方法评估DDI的具体考虑因素包括:
- DDI应该针对单个化合物进行表征,而不是针对治疗类药物(therapeutic classes of drugs),尽管例外情况是可能的(例如,如果相互作用机制是pH依赖性的,可以合并多个质子泵抑制剂)。
- 如果肇事者(perpetrators,犯罪药?)属于监管机构确定的同一类标志性的(Index)抑制剂/诱导剂(即弱,中等或强),则可以汇集多个肇事者(perpetrators,犯罪药?)以形成一个协变量类别。汇总的肇事者(perpetrators,犯罪药?)应该具有相同的相互作用机制,对其各自的代谢酶具有相似的特异性。
- 研究中应包括足够数量的伴随药物治疗的受试者。PK采样时间表应适当地表征感兴趣的PK参数。模拟可以确定在给定研究设计中检测限定量值的相互作用所需的受试者数量。
- 应对PK模型的所有生理上可信的结构元素(例如,清除率(CL / F),相对生物利用度(Frel),吸收速率等)进行相互作用。
Ⅳ.用于群体PK分析的数据
A.研究对象和协变量 确定数据是否足以解决预期的研究问题是任何群体PK分析中至关重要的一步(参见V.C部分)。数据集应包括足够数量的受试者,在信息性的时间点具有足够数量的PK样本。如果协变量分布较窄(对于连续协变量)或者该类别中的受试者数量不足(对于分类协变量),则不能声称协变量对药物暴露有影响或没有影响。许多连续协变量已经建立了定义类别级别的截止值。如果针对这些类别提出剂量推荐,则连续协变量的范围应优选地跨越整个类别而不仅仅是上端或下端。 B.PK采样方案
模型导出的PK参数的准确度和偏差取决于多个因素,包括受试者数量,每个受试者的样本数量和采样时间表。随着每个受试者的样品数量减少,PK样品的采样点时机的掌握的重要性增加。例如,如果分析的目的是匹配跨群体或剂型的Cmax观察,则应在吸收阶段收集足够数量的PK样品。鼓励申办方优质性地规划PK采样计划,以便群体PK模型获得最大的信息。 用于群体PK模型的优化设计的方法和可用软件在文献中的多个评论中被涵盖(Dodds,Hooker和Vicini 2005;
Nyberg等人。2015年; Ogungbenro和Aarons 2007; Ogungbenro和Aarons
2008)。下面的列表显示了一些采样计划示例。根据分析的目的,所列策略中的一个或组合可能就足够了: 患者被随机分配取样窗口,这些窗口是基于优质设计方法得出的。每个患者的样本数量和采样窗口的数量也是基于优质设计方法确定的。 - 患者随机提供两个或更多样本,当组合时,覆盖整个给药间隔。 - 大多数患者在指定的时间点提供一个样本,通常在下一次剂量之前。 鼓励申办方收集所有患者的PK数据。但是,每个患者的取样程度和取样患者的百分比最终应取决于数据的预期用途。例如,如果Cmax将用于后续的ER分析,那么应该在最大浓度(Tmax)的时间周围进行充分的采样(见第III.A.3节)。在任何情况下,重要的是要验证PK数据丢失的患者与其他患者没有差异。例如,缺乏PK数据的患者不应该是因为缺乏效果或不良事件而导致具有更高的脱落率。如果估计场合间(between-occasion)的变异性,则需要在不止一次的情况下每个人多个样本。忽略大的时间间差异可能会导致群体参数估计偏差(Karlsson和Sheiner
1993)。 Ⅴ. 数据分析
本文档的这个章节提供了群体PK分析方法学方面的一些指导原则。其他人已经描述了进一步的方法学考虑和良好实践,包括此处未涉及的主题(Ette和Williams
2007; Bonate和Steimer 2013; Mold和Upton 2013; Byon等人2013;
Tatarinova等人2013; Lunn等人。2002; Schmidt和Radivojevic 2014)。 A.数据初步审查
所有群体分析应首先检查观察到的数据。数据的初步检查使用图形和统计技术分离并揭示群体数据集中的模式和特征,并且可以提供强有力的诊断工具来确认假设,或者当不满足假设时,提出纠正措施(Tukey
1977; Ette和Ludden 1995)
)。例如,高度相关的协变量之间的相关性可能无法提供有关群体的独特信息。这种情况通常是例如由Cockcroft-Gault方程计算的体重和肌酐清除率的情况。应在群体PK分析报告中简要描述数据的相关初步检查。 B.模型开发
模型开发方法和优质实践建议不断发展。关于如何开发群体PK模型的具体建议超出了本指南的范围。然而,为了促进对群体PK模型的监管审查,申办方应明确描述其模型开发程序(有关群体PK报告的更多讨论,请参见第VII节)。下面提供了对监管审查很重要的模型开发的某些方面: 模型开发问题可以通过几种有效的方法来解决,每种方法都有其自身的优点和缺点。例如,可以基于几种方法或它们的可能组合(例如,逐步协变量分析,全协变量模型方法,Lasso)来进行协变量分析(Wählby,Jonsson和Karlsson
2002; Gastonguay 2004; Ribbing等人2007)。在这种情况下,申办方应证明为何使用特定方法。 - 可以基于生物学,生理学或异速缩放原理的当前知识建立协变量-参数关系。 - 关于缺失数据的问题,包括缺失协变量和低于量化限(LOQ)的数据,应采用适当的分析方法解决(Beal
2001; Bergstrand和Karlsson 2009; Johansson和Karlsson 2013;
Keiser等2015)。申办方应证明其在丢失数据和异常值方面的方法论方法,并提供敏感性分析。 - 申办方应区分异常(outlying)个体和异常数据点。在模型开发过程中,可以省略可疑异常值的各个数据点。但是,申办方应通过将最终模型重新设置为完整数据集来研究异常值对最终参数估计的影响。一般不鼓励移除可疑的异常(outlying)个体,除非异常的原因是违反方案或其他人为错误。申办方应指定如何在分析中识别和处理异常值。应在数据分析计划中预先指定将数据点声明为异常值的原因。例如,在某些情况下,加权残差大于5的数据点可被视为异常值。 C.模型验证 模型验证是任何群体PK分析的关键步骤,应该进行该步骤以检查所开发的模型是否能够充分表征观察到的数据和生成可靠的建模和仿真结果,以满足分析的需要。没有一种模型验证方法是通常足够评估模型的所有组件的。通常需要几种方法,以便每种方法的相对优势和弱点可以相互补充。通常,模型需要以可接受的偏差水平和可接受的准确度来描述数据。适当的模型应该是生物学合理的,与当前知识一致,并具有数学上可识别的参数。 模型验证取决于分析的目标,并应遵循适合目的的方法。在某些情况下,模型可能仅用于一个目的,但不适用于另一个目的。例如,CL/F收缩率高的模型可能无助于推导出用于连续ER分析的单个药物暴露水平(参见第III.A.3节)。然而,如果协变量建模方法对收缩不敏感,这种模型仍然可用于协变量分析。 向监管机构提交的材料应包含对所用模型验证方法的详细说明,并说明为何选择这些方法的原因(见第VII.A节)。 1.模型验证的常用方法
进行全面模型验证的程序不断发展,监管机构欢迎这一领域的创新。在文献中报道了几种常用的模型验证方法(Karlsson和Savic 2007; Byon等人2013)。其中一些方法将在下面的部分中讨论。 基础的拟合优度(GOF)图说明了模型描述观测数据的程度。GOF图还用于评估模型假设(例如随机效应的正态性)并指导模型开发。尽管GOF图可以显示整体拟合是可接受的,但是通常需要对患者亚组中的模型进行额外评估。例如,如果该模型将用于预测儿科患者的药物暴露,则应对所有年龄组进行模型验证。对重要患者特征(例如,按年龄组或CYP多态性分层),研究设计(例如,剂量或制剂)或其他重要变量进行分层的GOF图,通常比完整的数据集GOF图,有足够的表现(adequate
performance)更据说服力。 以下是一些被认为信息丰富的GOF图表: - 因变量(DV)与个体预测(IPRED) - DV与群体预测(PRED) - 绝对个体加权残差(|IWRES|)与IPRED或时间(时间应评估:连续时间IVAR和剂量后的时间TAD。) - 条件加权残差(CWRES)与PRED或时间 - IPRED,PRED和观察与时间的代表性样本(每个受试者一个图) - 随机效应的直方图或分位数-分位数(QQ)图 - 随机效应之间的相关性 - 随机效应与协变量(在包含协变量之前和之后比较时,该图最具信息性。) 个别参数,IPRED和IWRES,往往会在观察较少的个体中朝着群体估计收缩。当收缩率很高(通常大于20-30%)时,依赖于EBE,IPRED或IWRES的诊断图可能变得无法提供信息,随机效应和协变量之间的相关性可能会变得模糊(Savic和Karlsson
2009)。此外,高收缩率可能会限制使用ER分析的个别事后估计值(见第III.A.3节)。基于模拟的诊断图不会以类似的方式受到收缩的影响,并且在收缩率高时可以为诊断目的提供更多信息。 有几种可用的基于仿真的诊断,包括但不限于视觉预测检验(VPC),预测校正的VPC(pcVPC)和数字预测检查(NPC)(Bergstrand等人,2011)。 GOF标准也可以通过一些数值度量来反映,例如参数精度的估计。参数精度的估计可以提供关于支持该参数的数据充足性的有价值信息。参数不确定性可以通过几种方法估算,包括自举过程(bootstrap
procedures),对数似然性分析,或使用参数估计的渐近标准误差。除参数不确定性外,重要的是将参数点估计值与先前的分析进行比较,并评估点估计值的生理合理性。评估模型的另一个有用的数值度量是条件值。条件数(最大和最小特征值的比率)超过1000表示观测数据不能支持一个或多个参数的估计(Montgomery,Vining和Peck
2012)。随着附加数据的出现,可以简化和更新过度参数化的模型。 可以根据一组内部或外部测试数据检查模型的性能。上面提到的验证方法依赖于用于模型构建的数据,因此被视为内部验证。另一方面,外部验证依赖于未用于模型构建的数据。药物开发计划中收集的数据可以分为模型建筑数据集和测试数据集。测试数据集通常用于外部验证。数据拆分是一种强大的模型评估方法。但是,在决定采用数据分割方法之前,申办方应考虑数据丢失对模型检测协变量关系的能力的潜在影响,并以可接受的精度估算参数。 当模型的目的是模拟超出临床研究范围的场景的药代动力学特征时,还有额外的不确定性。这种不确定性可以通过对参数估计及其对用于决策的指标的影响的敏感性分析来解决(Kimko和Peck,2010)。 D.基于群体PK模型的模拟
模拟应该基于概述要执行的模拟研究的方案。方案中的详细程度应与模拟所解决的问题的复杂性和影响相对应。应验证用于模拟的模型,以解决模拟研究试图回答的具体问题。虽然模型以前可以使用和验证,但如果新目的与原始目的不同,则需要重新验证。根据模型的目的,可以在模拟中添加各种级别的不确定性和随机变异(uncertainty
and variability)。下面讨论一些例子。
1.基于固定效应估计的模拟
在简单的形式中,模拟用于说明典型个体的药物浓度曲线。这种模拟基于固定效应参数的典型估计。请注意,基于典型参数估计的预测与平均预测不对应。平均预测通过基于具有群体PK参数个体间随机变异和残差的模拟,计算平均浓度-时间曲线来获得。 2.基于固定效应估计不确定性的仿真
如果希望说明典型受试者的药物暴露达到或保持在特定截止点以上或者如果希望说明协变量的影响的概率,则可以考虑参数估计的不确定性。例如,可以基于具有固定效应参数的不确定性的模拟来生成说明协变量对AUC或其他参数的影响的森林图,从而有助于解释协变量对曝光的相对重要性。这些类型的模拟也可用于评估新剂量方案在未来试验中的性能。另外,具有参数不确定性的模拟可用于图形化地说明参数准确度对PK曲线的影响。 3.基于受试者间变异性估计的模拟
当目的是显示研究群体中个体预测浓度的范围时,在模拟中考虑PK参数中的受试者间变异性(BSV)。当观察到的浓度范围是主要关注点时,将残余误差添加到个体预测中。 如果目的是预测未来群体的观测浓度范围,那么除了残差和BSV之外,还应考虑固定效应参数的不确定性。 应考虑随机效应之间的相关性,以避免受试者中不切实际的参数组合。考虑BSV并包括协变量效应的模拟应该在具有实际群体统计变量的群体中进行。群体统计学变量可以从数据库中获得,通过重新取样原始研究中的个体或通过对协变量分布及其在目标群体中的相关性进行采样而生成。 Ⅵ. 基于群体PK分析结果的药品说明书
群体PK分析的结果可以在临床药物分析部分中呈现,并在适当时在药品说明书的其他部分中总结。其他相关药品说明书部分不应重复此详细信息,而应根据这些结果提供简洁的描述或建议,然后酌情交叉引用临床药物部分。通常,没有必要明确声明信息是基于群体PK分析。关于开发临床药理学部分的建议在FDA工业指南中讨论,该指南题为“人类处方药和生物制品药品说明书的临床药理学部分-内容和格式”。 Ⅶ. 群体PK研究报告
本节概述了申办方提交的群体PK分析的推荐格式和内容,以支持其药物的临床药理学计划。群体PK分析可适用于药物开发过程中的多个点,例如在研究性新药申请(IND),NDA或上市后阶段。由于临床数据的可用性或质量,每个阶段的群体PK分析的深度和广度可以变化。 对于监管决策很重要的群体PK研究报告应放在在电子格式通用技术文件(eCTD)的模块5.3.3.5,相应程序,数据集和定义文件应放置在eCTD模块5数据集文件夹下。 A.群体PK报告的格式和内容
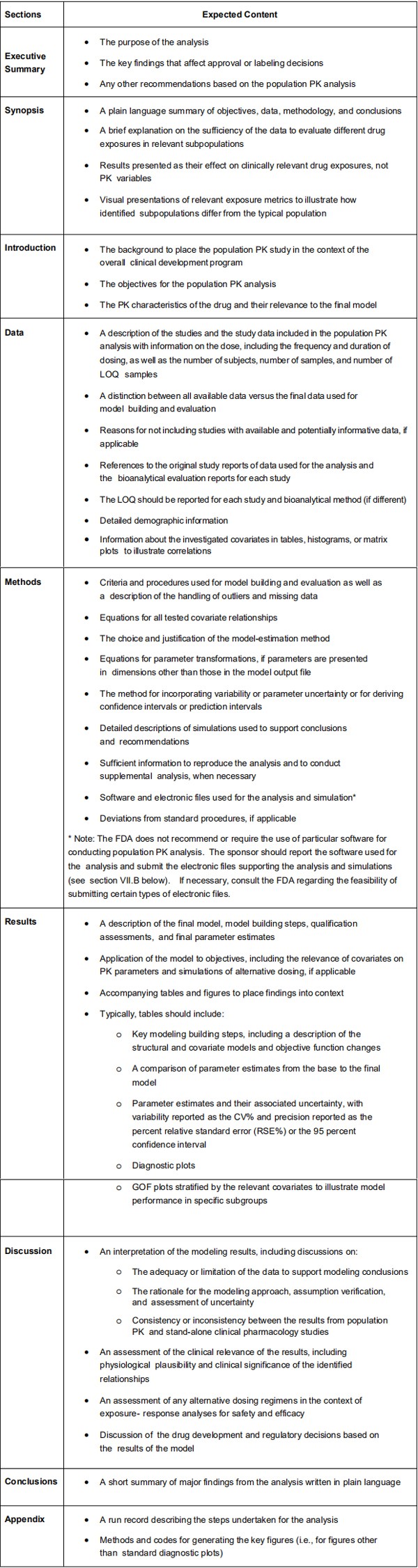
为了能够对群体PK分析进行有效和一致的审查,FDA建议群体PK分析的结果应该附有结构化群体PK报告。报告应包含以下部分:(1)执行摘要,(2)概要,(3)介绍,(4)数据,(5)方法,(6)结果,(7)讨论,(8)结论,(9)附录(如适用)。表1中简要讨论了包含在这些部分中的预期信息/要素。包含基于群体PK分析信息的带注释标签(annotated
labeling)的部分应包括群体PK报告的超链接。关于报告的其他讨论可在科学文献中找到(Dykstra等人,2015)。 表1:群体PK研究报告各部分的预期内容
表1:群体PK研究报告各部分的预期内容(原文) B.向机构提交电子文件 申办方应参考FDA网站10,获取有关提交数据和相关文件(例如编码脚本)的一般建议。至关重要的是,为基础,最终和关键中间模型提交的所有数据集和模型文件与用于在报告附录中生成模型输出的数据集和模型文件相同。此外,FDA工作人员应该能够识别使用输出文件构建的任何数据集的源数据或来自群体PK分析的后处理结果。例如,如果来自群体PK分析的暴露指标包含在E-R分析的数据集中,则申办方应确保群体PK模型输出与构建数据集所涉及的任何后处理步骤之间的可追溯性。这可以通过提供定义文件,审阅者指南或用于数据集组装的代码来完成。 监管机构PK分析得出的所有结论都应由监管机构以现有的代码和数据重现。在群体PK数据集中包括每个受试者的受试者标识符信息并确保各个临床研究报告数据集中的标识符相同也是重要的。如果需要在群体PK模型生成的个体水平输出(例如CL或Vd的个体,事后估计)与来自个体临床研究报告的疗效或安全数据集之间进行数据整合,则此信息至关重要。 Ⅷ. 专业术语词汇表 Ⅸ. 参考文献 (文章名称翻译后的列表) Barbour,AM,MJ Fossler和J Barrett,2014年,儿童患者剂量选择的实际考虑因素,以确保目标暴露要求,AAPS J,16(4):749-55。 Beal,SL,2001,如何使用低于定量限的某些数据拟合PK模型,J Pharmacokinet Pharmacodyn,28(5):481-504。 Bergstrand,M,AC Hooker,JE Wallin和MO Karlsson,2011,用于诊断非线性混合效应模型的预测校正视觉预测检验,AAPS J,13(2):143-51。 Bergstrand,M和MO Karlsson,2009,处理混合效应模型中低于定量下限的数据,AAPS J,11(2):371-80。 Bhattaram,VA,BP Booth,RP Ramchandani,BN Beasley,Y Wang,V Tandon,JZ
Duan,RK Baweja,PJ Marroum和RS
Uppoor,2005,Pharmacometrics对药物批准和药品说明书决策的影响:对42种新药的调查应用,AAPS
J,7(3):E503-E512。 Bonate,PL和JL Steimer,2013,Pharmacokinetic-Pharmacodynamic Modeling and Simulation,Springer。 Bonate,PL,M Ahamadi,N Budha,A de la Pena,JC Earp,Y Hong,MO
Karlsson,P Ravva,A Ruiz-Garcia,H Struemper,JR Wade,2016,评估不受控制的药物 -
药物相互作用的方法和策略在群体药代动力学分析中:来自国际药理学会(ISOP)工作组的结果,J Pharmacokinet
Pharmacodyn,43(2):123-35。 Byon,W,MK Smith,P Chan,MA Tortorici,S Riley,H Dai,J Dong,A Ruiz -
Garcia,K Sweeney and C Cronenberger,2013,进行群体建模的优质实践和指南:内部经验
群体药代动力学分析指南,CPT Pharmacometrics Syst Pharmacol,2(7):1-8。 Dodds,MG,AC Hooker和P Vicini,2005,健壮的群体药代动力学实验设计,J Pharmacokinet Pharmacodyn,32(1):33-64。 Dykstra,K,N Mehrotra,CWTornøe,H Kastrissios,B Patel,N Al - Huniti,P Jadhav,Y Wang和W Byon,2015,群体药代动力学分析报告指南,J Pharmacokinet Pharmacodyn,42(3):301-14。 Ette,EI和TM Ludden,1995,群体药代动力学建模:信息图形的信息,Pharma Res,12(12):1845-55。 Ette,EI和PJ Williams,2007,定量药理学:定量的药理学,John Wiley&Sons。 Gastonguay,MR,2004,协变量效应的全模型估计方法:基于临床重要性和估计精度的推断,J AAPS,6(S1):W4354。 Grasela Jr,TH和LB Sheiner,1991,观察数据的药物统计学建模,J Pharmacokinet Biopharm,19(S3):S25-S36。 Holford,N,YA Heo和B Anderson,2013,婴儿和成人的药代动力学标准,J Pharmaceut Sci,102(9):2941-52。 Johansson,ÅM和MO Karlsson,2013,处理遗漏协变量数据的方法比较,J AAPS,15(4):1232-41。 Karlsson,M和RM Savic,2007,进行模型诊断所用的诊断,Clin Pharmacol Ther,82(1):17-20。 Karlsson,MO和LB Sheiner,1993,“群体药代动力学分析中模拟相互作用变异性的重要性”,J Pharmacokinet Biopharm,21(6):735-50。 Keiser,RJ,RS Jansen,H Rosing,B Thijssen,JH Beijnen,JHM Schellens和ADR
Huitema,2015,浓度数据的合并低于群体药代动力学分析中的定量限,Pharmacol Res
Perspect,3(2):e00131。 Kimko,HHC和CC Peck,2010年,临床试验模拟和定量药理学:应用和趋势,Springer Science & Business Media. Lee,JY,CE Garnett,JVS Gobburu,VA Bhattaram,S Brar,JC Earp,PR
Jadhav,K Krudys,LJ Lesko和F Li,2011,定量药理学对新药上市申请和药品说明书的决定的影响,50
(10):627-35。 Lunn,DJ,N Best,A Thomas,J Wakefield和D Spiegelhalter,2002,群体PK /
PD模型的贝叶斯分析:一般概念和软件,J Pharmacokinet Pharmacodyn,29(3):271-307。 Mahmoood I.儿童剂量:对儿科药物开发和临床环境中药代动力学异速生长标度和建模方法的批判性回顾。Clin Pharmacokinet 2014; 53:327-46。 Marshall, SF, R Burghaus, V Cosson, SYA Cheung, M Chenel, O
DellaPasqua, N Frey, B Hamren, L Harnisch, and F Ivanow,
2015,模型知情的药物发现和开发(MID3)的良好实践:实践,应用和文档,CPT:Pharmacometrics Syst
Pharmacol,5(3):93-122。 Montgomery,DC,GG Vining和EA Peck,2012,线性回归分析导论,Wiley。 Mold,DR和RN Upton,2013,群体建模,模拟和基于模型的药物开发的基本概念 - 第2部分:药代动力学建模方法简介,CPT:Pharmacometrics Syst Pharmacol,2(4):e38。 Nyberg,J,C Bazzoli,K Ogungbenro,A Aliev,S Leonov,S Duffull,AC
Hooker和FMentré,2015,用于群体药代动力学 - 药效学研究中的设计评估的方法和软件工具,Br J Clin
Pharmacol,79(1 ):6-17。 Ogungbenro,K和L Aarons,2007,使用现有信息设计群体药代动力学实验,Xenobiotica,37(10-11):1311-30。 Ogungbenro,K和L Aarons,2008,用于群体药代动力学实验的采样窗口设计的优化,J Pharmacokinet Pharmacodyn,35(4):465-82。 Ribbing,J,J Nyberg,O Caster和EN Jonsson,2007,用于非线性混合效应模型中的预测协变量模型构建的Lasso-A新方法,J Pharmacokinet Pharmacodyn,34(4):485-517。 Savic,RM和MO Karlsson,2009,诊断经验贝叶斯估计收缩的重要性:问题和解决方案,J AAPS,11(3):558-69。 Schmidt,H和A Radivojevic,2014,使用综合工作流增强群体药代动力学建模效率和质量,J Pharmacokinet Pharmacodyn,41(4):319-34。 Sheiner,LB,B Rosenberg和VV Marathe,1977,从常规临床数据估计药代动力学参数的群体特征,J Pharmacokinet Biopharm,5(5):445-79。 Svensson,EM,C Acharya,B Clauson,KE Dooley和MO Karlsson,2016,药物动力学相互作用的长期半衰期药物 - 需要基于模型的分析证据,J AAPS,18(1):171- 79。 Tatarinova,T,M Neely,J Bartroff,M van Guilder,W Yamada,D Bayard,R
Jelliffe,R Leary,A Chubatiuk和A
Schumitzky,2013,两种一般方法用于群体药代动力学建模:非参数自适应网格和非参数贝叶斯,J Pharmacokinet
Pharmacodyn,40(2):189-99。 Tukey,JW,1977,探索性数据分析,Addison-Wesley。 Wählby,U,MR Bouw,EN Jonsson和MO Karlsson,2002,NONMEM中统计子模型的I型误差率的评估,J Pharmacokinet Pharmacodyn,29(3):25-69。 Wählby,U,EN Jonsson和MO Karlsson,2001,NONMEM中协变量效应的实际显着性水平的评估,J Pharmacokinet Pharmacodyn,28(3):231-52。 Wählby,U,EN Jonsson和MO Karlsson,2002,逐步协变量模型构建策略在群体药代动力学 - 药效学分析中的比较,AAPS PharmSci,4(4):68-79。 Wang,Y,PR Jadhav,M Lala和JV Gobburu,2012,关于在设计儿科药代动力学研究时得出样本大小的准确标准的澄清,J Clin Pharm,52(10):1601-06。 Wang,DD,Y Yu,N Kassir,M Zhu,WD Hanley,JC Earp,AT Chow,M Gupta,C
Hu,2017,药物 - 药物相互作用评估中的群体方法的应用:模拟评估,J Clin Pharmacol ,57(10):1268-1278。 Zhang,Y,X Wei,G Bajaj,JS Barrett,B Meibohm,A Joshi和M Gupta,2015,儿科患者治疗性蛋白质开发的挑战和考虑,J Clin Pharmacol,55(S3):S103-15。 FDA发布的行业指导原则: 2019年题为“ 评估食品对INDs和NDAs中药物的影响 - 临床药理学考虑因素”的指南草案 2003年最终指南题为“肝功能受损患者的药代动力学 - 研究设计,数据分析以及对剂量和标记的影响” 2010年指南题为“ 肾功能受损患者的药代动力学 - 研究设计,数据分析以及对剂量和标签的影响” 2017年题为“ 体外代谢 - 和转运蛋白介导的药物 - 药物相互作用研究”的指南草案 2017年题为“ 临床药物相互作用研究 - 研究设计,数据分析和临床意义”的指南草案 参考文献(文章名称未翻译的原始列表) Barbour, AM, MJ Fossler, and J Barrett, 2014, Practical
Considerations for Dose Selection in Pediatric Patients to Ensure Target
Exposure Requirements, AAPS J, 16(4):749-55. Beal, SL, 2001, Ways to Fit a PK Model With Some Data Below the
Quantification Limit, J Pharmacokinet Pharmacodyn, 28(5):481-504. Bergstrand, M, AC Hooker, JE Wallin, and MO Karlsson, 2011,
Prediction-Corrected Visual Predictive Checks for Diagnosing Nonlinear
Mixed-Effects Models, AAPS J, 13(2):143- 51. Bergstrand, M and MO Karlsson, 2009, Handling Data Below the Limit
of Quantification in Mixed Effect Models, AAPS J, 11(2):371-80. Bhattaram, VA, BP Booth, RP Ramchandani, BN Beasley, Y Wang, V
Tandon, JZ Duan, RK Baweja, PJ Marroum, and RS Uppoor, 2005, Impact of
Pharmacometrics on Drug Approval and Labeling Decisions: A Survey of 42
New Drug Applications, AAPS J, 7(3):E503-E512. Bonate, PL and JL Steimer, 2013, Pharmacokinetic-Pharmacodynamic Modeling and Simulation, Springer. Bonate, PL, M Ahamadi, N Budha, A de la Pena, JC Earp, Y Hong, MO
Karlsson, P Ravva, A Ruiz-Garcia, H Struemper, JR Wade, 2016, Methods
and Strategies for Assessing Uncontrolled Drug-Drug Interactions in
Population Pharmacokinetic Analyses: Results from the International
Society of Pharmacometrics (ISOP) Working Group, J Pharmacokinet
Pharmacodyn, 43(2):123-35. Byon, W, MK Smith, P Chan, MA Tortorici, S Riley, H Dai, J Dong, A
Ruiz‐Garcia, K Sweeney, and C Cronenberger, 2013, Establishing Best
Practices and Guidance in Population Modeling: An Experience With an
Internal Population Pharmacokinetic Analysis Guidance, CPT
Pharmacometrics Syst Pharmacol, 2(7):1-8. Dodds, MG, AC Hooker, and P Vicini, 2005, Robust Population
Pharmacokinetic Experiment Design, J Pharmacokinet Pharmacodyn,
32(1):33-64. Dykstra, K, N Mehrotra, CW Tornøe, H Kastrissios, B Patel, N
Al‐Huniti, P Jadhav, Y Wang, and W Byon, 2015, Reporting Guidelines for
Population Pharmacokinetic Analyses, J harmacokinet Pharmacodyn,
42(3):301-14. Ette, EI and TM Ludden,1995, Population Pharmacokinetic Modeling:
The Importance of Informative Graphics, Pharma Res, 12(12):1845-55. Ette, EI and PJ Williams, 2007, Pharmacometrics: The Science of Quantitative Pharmacology, John Wiley & Sons. Gastonguay, MR, 2004, A Full Model Estimation Approach for Covariate
Effects: Inference Based on Clinical Importance and Estimation
Precision, J AAPS, 6(S1):W4354. Grasela Jr, TH and LB Sheiner, 1991, Pharmacostatistical Modeling
for Observational Data, J Pharmacokinet Biopharm, 19(S3):S25-S36. Holford, N, YA Heo, and B Anderson, 2013, A Pharmacokinetic Standard
for Babies and Adults, J Pharmaceut Sci, 102(9):2941-52. Johansson, ÅM and MO Karlsson, 2013, Comparison of Methods for Handling Missing Covariate Data, J AAPS, 15(4):1232-41. Karlsson, M and RM Savic, 2007, Diagnosing Model Diagnostics, Clin Pharmacol Ther, 82(1):17-20. Karlsson, MO and LB Sheiner, 1993, The Importance of Modeling
Interoccasion Variability in Population Pharmacokinetic Analyses, J
Pharmacokinet Biopharm, 21(6):735-50. Keizer, RJ, RS Jansen, H Rosing, B Thijssen, JH Beijnen, JHM
Schellens, and ADR Huitema, 2015, Incorporation of Concentration Data
Below the Limit of Quantification in Population Pharmacokinetic
Analyses, Pharmacol Res Perspect, 3(2):e00131. Kimko, HHC and CC Peck, 2010, Clinical Trial Simulation and
Quantitative Pharmacology: Applications and Trends, Springer Science
& Business Media. Lee, JY, CE Garnett, JVS Gobburu, VA Bhattaram, S Brar, JC Earp,
PR Jadhav, K Krudys, LJ Lesko, and F Li, 2011, Impact of Pharmacometric
Analyses on New Drug Approval and Labelling Decisions, Clin
Pharmacokinet, 50(10):627-35. Lunn, DJ, N Best, A Thomas, J Wakefield, and D Spiegelhalter, 2002,
Bayesian Analysis of Population PK/PD Models: General Concepts and
Software, J Pharmacokinet Pharmacodyn, 29(3):271-307. Mahmoood I. Dosing in Children: a Critical Review of the
Pharmacokinetic Allometric Scaling and Modelling Approaches in
Paediatric Drug Development and Clinical Settings. Clin Pharmacokinet
2014; 53: 327-46. Marshall, SF, R Burghaus, V Cosson, SYA Cheung, M Chenel, O
DellaPasqua, N Frey, B Hamren, L Harnisch, and F Ivanow, 2015, Good
Practices in Model‐Informed Drug Discovery and Development (MID3):
Practice, Application and Documentation, CPT: Pharmacometrics Syst
Pharmacol, 5(3):93-122. Montgomery, DC, GG Vining, and EA Peck, 2012, Introduction to Linear Regression Analysis, Wiley. Mould, DR and RN Upton, 2013, Basic Concepts in Population Modeling,
Simulation, and Model-Based Drug Development—Part 2: Introduction to
Pharmacokinetic Modeling Methods, CPT: Pharmacometrics Syst Pharmacol,
2(4):e38. Nyberg, J, C Bazzoli, K Ogungbenro, A Aliev, S Leonov, S Duffull, AC
Hooker, and F Mentré, 2015, Methods and Software Tools for Design
Evaluation in Population Pharmacokinetics–Pharmacodynamics Studies, Br J
Clin Pharmacol, 79(1):6-17. Ogungbenro, K and L Aarons, 2007, Design of Population
Pharmacokinetic Experiments Using Prior Information, Xenobiotica,
37(10-11):1311-30. Ogungbenro, K and L Aarons, 2008, Optimisation of Sampling Windows
Design for Population Pharmacokinetic Experiments, J Pharmacokinet
Pharmacodyn, 35(4):465-82. Ribbing, J, J Nyberg, O Caster, and EN Jonsson, 2007, The Lasso—A
Novel Method for Predictive Covariate Model Building in Nonlinear Mixed
Effects Models, J Pharmacokinet Pharmacodyn, 34(4):485-517. Savic, RM and MO Karlsson, 2009, Importance of Shrinkage in
Empirical Bayes Estimates for Diagnostics: Problems and Solutions, J
AAPS, 11(3):558-69. Schmidt, H and A Radivojevic, 2014, Enhancing Population
Pharmacokinetic Modeling Efficiency and Quality Using an Integrated
Workflow, J Pharmacokinet Pharmacodyn, 41(4):319-34. Sheiner, LB, B Rosenberg, and VV Marathe, 1977, Estimation of
Population Characteristics of Pharmacokinetic Parameters From Routine
Clinical Data, J Pharmacokinet Biopharm, 5(5):445-79. Svensson, EM, C Acharya, B Clauson, KE Dooley, and MO Karlsson,
2016, Pharmacokinetic Interactions for Drugs with a Long
Half-Life—Evidence for the Need of Model-Based Analysis, J AAPS,
18(1):171-79. Tatarinova, T, M Neely, J Bartroff, M van Guilder, W Yamada, D
Bayard, R Jelliffe, R Leary, A Chubatiuk, and A Schumitzky, 2013, Two
General Methods for Population Pharmacokinetic Modeling: Non-Parametric
Adaptive Grid and Non-Parametric Bayesian, J Pharmacokinet Pharmacodyn,
40(2):189-99. Tukey, JW, 1977, Exploratory Data Analysis, Addison-Wesley. Wählby, U, MR Bouw, EN Jonsson, and MO Karlsson, 2002, Assessment of
Type I Error Rates for the Statistical Sub-Model in NONMEM, J
Pharmacokinet Pharmacodyn, 29(3):25-69. Wählby, U, EN Jonsson and MO Karlsson, 2001, Assessment of Actual
Significance Levels for Covariate Effects in NONMEM, J Pharmacokinet
Pharmacodyn, 28(3):231-52. Wählby, U, EN Jonsson and MO Karlsson, 2002, Comparison of Stepwise
Covariate Model Building Strategies in Population
Pharmacokinetic-Pharmacodynamic Analysis, AAPS PharmSci, 4(4):68-79. Wang, Y, PR Jadhav, M Lala, and JV Gobburu, 2012, Clarification on
Precision Criteria to Derive Sample Size When Designing Pediatric
Pharmacokinetic Studies, J Clin Pharm, 52(10):1601-06. Wang, DD, Y Yu, N Kassir, M Zhu, WD Hanley, JC Earp, AT Chow, M
Gupta, C Hu, 2017, The Utility of a Population Approach in Drug-Drug
Interaction Assessments: A Simulation Evaluation, J Clin Pharmacol,
57(10):1268-1278. Zhang, Y, X Wei, G Bajaj, JS Barrett, B Meibohm, A Joshi, and M
Gupta, 2015, Challenges and Considerations for Development of
Therapeutic Proteins in Pediatric Patients, J Clin Pharmacol,
55(S3):S103-15. FDA guidances for industry: 2019 draft guidance entitled Assessing the Effects of Food on Drugs
in INDs and NDAs — Clinical Pharmacology Considerations11 2003 final guidance entitled Pharmacokinetics in Patients With
Impaired Hepatic Function — Study Design, Data Analysis, and Impact on
Dosing and Labeling 2010 draft guidance entitled Pharmacokinetics in Patients With
Impaired Renal Function — Study Design, Data Analysis, and Impact on
Dosing and Labeling10 2017 draft guidance entitled In Vitro Metabolism- and Transporter-Mediated Drug-Drug Interaction Studies10 2017 draft guidance entitled Clinical Drug Interaction Studies—Study Design, Data Analysis, and Clinical Implications10 英文原文下载链接: https://www.fda.gov/media/128793/download