
前言:
2021年8月份发表在Pathogens杂志的一篇文章,采用CLC进行了HPV病毒基因组进化和发病机制研究。标题为“HPV DeepSeq:一种使用自动化工作流和CLC Genomics Workbench中定制的乳头状瘤病毒数据库进行NGS数据分析和可视化的超快速方法”。该文章为临床病毒学的研究提供了一套可参考的分析思路,非常客观的评估了CLC在病毒基因组分析中的优势:
1. 整合了临床相关和科学分类的HPV参考基因组序列;
2. 提供一站式的分析流程,包括基因组mapping、基因组拼接、taxonomic profiling、alpha & beta diversity分析、统计学差异分析等;
3. 结果可视化。
本文将详细解读如何使用凯杰CLC软件搭建分析流程进行HPV病毒基因组进化和发病机制的研究。
研究背景:
HPV(人类乳头瘤病毒)是全球第二大主要癌症传染源,导致每年全球新增570000例宫颈癌和120000例其他肛门生殖道和口咽癌病例。HPV是一种小型(8000碱基对)双链环状DNA病毒,其基因组在进化过程中获得了癌基因E6和E7以及后来的E5。这些癌基因的遗传差异赋予了病毒不同的表型和致癌潜力。最近的系统发育分析还表明,不同HPV基因型的解剖部位偏好和组织取向可能是病毒生态位适应宿主生态系统的结果。位点特异性基因型和病毒组组成可能进一步由宿主的免疫反应决定。因此,解剖病毒组特征对于我们理解作为发病机制基础的“生态位特异性病毒-宿主适应性进化”至关重要。
HPV亚基因组的分类和可视化
使用的CLC工作流:Data quality control (QC) and taxonomic profiling
该工作流生成了下图的丰度图,展示了识别出的分类群的名称、7级分类命名法、覆盖率估计和丰度值(即在与分类群相关的样本中找到的原始或相对读取序列数量)。

研究使用了Sanger和NGS测序技术检测每个样本中的HPV基因型和亚谱系。Sanger测序分析了每个样本中的单一显性HPV基因型。NGS在检测混合基因型(截止值六种基因型)和低丰度基因型(截止值≥总成分的1%)上具有更高的分辨率。
LSIL/HSIL HPV群体的多样性分析和可视化
使用的CLC工作流:Merge and Estimate Alpha and Beta Diversities
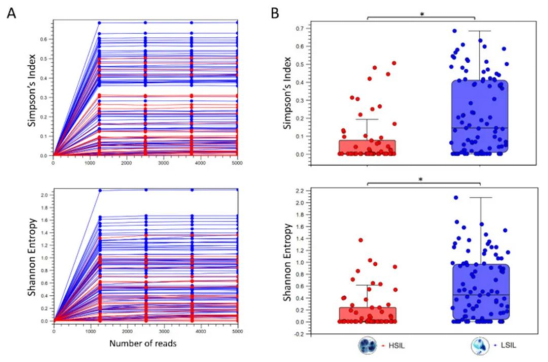
Alpha多样性是通过对每个样本中不同深度(读取序列的数量)的丰度进行亚采样来计算的。稀疏分析参数定义了Alpha多样性曲线的粒度,如下图A所示。通过使用丰度表元数据(即用于聚合样本组 “LSIL低度鳞状上皮内病变”和“HSIL高度鳞状上皮内病变” 的元数据),生成了下图B中的Mann-Whitney U统计结果。
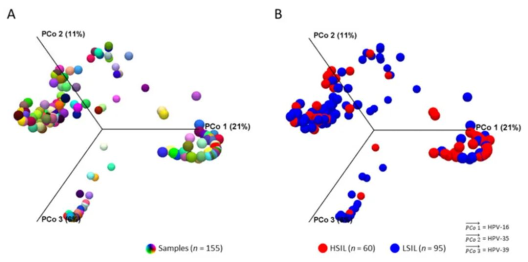
Beta多样性分析工作流程输出的是样本和二维或三维PCoA图之间的Bray Curtis距离矩阵。3D PCoA图直观地显示了所有样本之间HPV成分的差异(下图A)。对HSIL和LSIL样本进行分组后,不同的HPV群体和影响力较大的基因型在可视化结果上展示出明显的区分(下图B)。
LSIL/HSIL HPV群体的差异丰度分析和可视化
使用的CLC工作流:Convert Abundance Table to Experiment和Proportion-based Statistical Analysis
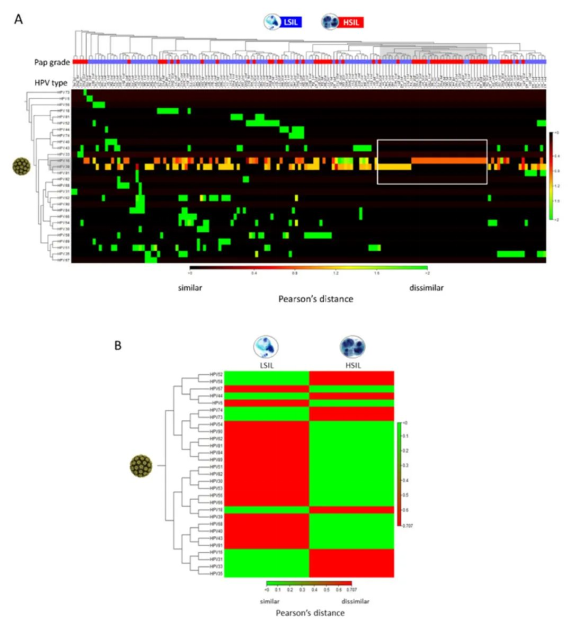
具有HPV基因型的HSIL和LSIL样本在蓝-红谱中具有相似的高丰度(~100%的读取序列),而在红谱中丰度不同(下图A)。LSIL和HSIL样本的热图显示了不同的HPV谱,其主要基因型显示为红色(下图B)。
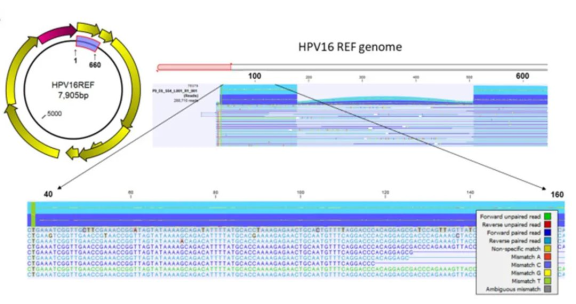
读取序列比对及比对轨迹的可视化
使用的CLC工作流:Map Reads to Reference
下图为一个代表性的比对轨迹,放大即可使序列可视化到核苷酸水平,以便与参考基因组进行比较并检测变异。
总结
此研究使用CLC工作流和分析工具从深度测序的临床样本中分析了HPV病毒组。CLC高效快速地完成了HPV亚基因组的分类和可视化,揭示了LSIL和HSIL病毒群落之间的差异。Alpha和Beta多样性分析不到5秒即可完成,LSIL/HSIL HPV群体的差异丰度分析和热图可视化在几秒钟内即可完成,处理时间与样本中合并序列的数量呈线性相关。NGS和分类学分析的HPV基因分型结果得到了Sanger和BLAST结果的验证。NGS技术提供了更丰富的病毒组和疾病状态(即从LSIL到HSIL)间进化动力学的信息。
作者在文章中提到,最近发表的另一篇文章中采用了9个基于命令行的生物信息学工具才能完成这些分析,而对于临床病毒学家和医学科学家,学习命令行需要花费大量的时间成本。可见生信分析软件对于科研工作的高效推进十分重要。


