
鉴别瘤牛特有的外显子组变异,这些变异不存在野牛与牛的基因组中(Cosart et al. 2011)。此外,我们使用CLC Genomics Workbench内置工具gene ontology,试图将瘤牛特有的序列变异与改变的通路联系起来。
数据:
1.所有三种牛的短测序序列
2.牛参考序列、牛基因注释和来自dbSNP / Ensembl的牛变异数据
3.牛基因组GO注释文件
软件:CLC Genomics Workbench 7.0
分析流程:
1.准备数据集
使用CLC Genomics Workbench自动下载基因组的工具下载具有注释的牛基因组,选择Illumina格式导入测序数据。选择标准导入工具导入牛注释文件,即Gene Ontology注释文件。在进行Read Mapping之前,创建测序数据QC报告(默认参数),评估测序数据是否需要修剪。根据报告内容判断,这些测序数据不需要修剪,可以直接用于Read Mapping。
2.测序数据比对和变异检测
首先,进行Read Mapping分析,三种牛测序数据分别与对应的参考序列比对(默认参数),生成Track和分析报告结果。然后,使用Local Realignment工具本地重新比对Mappings,无指导Track。最后,使用Probabilistic Variant Detection Caller工具(默认参数)分析Mappings的SNPs、小片段插入与缺失。
3.注释变异
选择Known Variants tools的Annotate工具,可以为变异添加其他信息。
4.筛选变异
我们对瘤牛特有的变异感兴趣,需要从鉴别的变异筛选出符合要求的变异。只要有变异Track,我们便可以使用Filter Against Know Variants 工具。我们将瘤牛设置为靶标Track,野牛与牛被设置为已知Tracks,变异不存在已知Tracks中。另外,我们可以将野牛与牛的Mappings作为对照,筛选瘤牛特有的变异。
进而,我们选择只出现在编码区域的变异,这个步骤根据Track 工具、CDS track与参考序列重叠的部分进行。然后,使用Amino Acid Changes工具筛选导致氨基酸变化的变异,这个步骤通过选择导致编码区域的非同义改变,进一步减少变异数量。
5.过表达分析
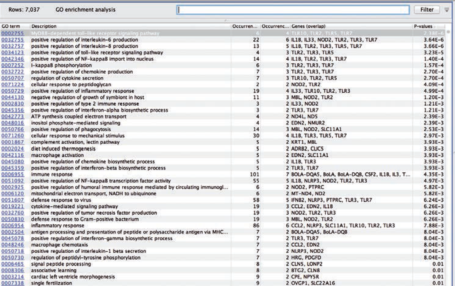
分析瘤牛特有的变异对应的基因具有哪些共同点,这表示瘤牛具有一些改变的通路。如果瘤牛特有的变异在任意一个生物学通路中过表达,我们便使用GO Enrichment Analysis工具进行分析。参考序列Track需要指定基因名和GO track。GO富集分析结果表,每行可以使用计算的P值分类,P值是根据比较在一个样本中出现的频率与在所有基因中出现的频率的几何分布测试计算的。
结果:
三种牛的测序数据比对结果显示,超过99%的测序数据能够成功比对。变异分析发现牛与瘤牛的基因组中大约具有38,000个变异,野牛基因组中大约具有72,000个变异。将野牛与牛的Read Mappings作为对照,筛选到瘤牛特有的变异是4733个,进一步根据变异是否存在编码区和是否导致氨基酸改变这两个条件,最终筛选到459个变异。
GO富集分析结果表根据P值分类,最上方几行的通路与免疫反应相关(见表1)。该结果与之前的发现相匹配,都显示瘤牛对一些感染的免疫反应,与野牛和牛类的不同。与其他两种物种相比,瘤牛还具有更好的天生免疫应答(Freeman et al. 2008)。
表1 GO富集分析结果,以P值升序排列,最上方的内容被突出显示,对应的变异选择见图1
Workflow实现自动分析数据
上文陈述的大多数步骤可以组合成Workflows(见图1),这些分析流程由批量模式操作组成,极大地简化了分析,能够重复操作。
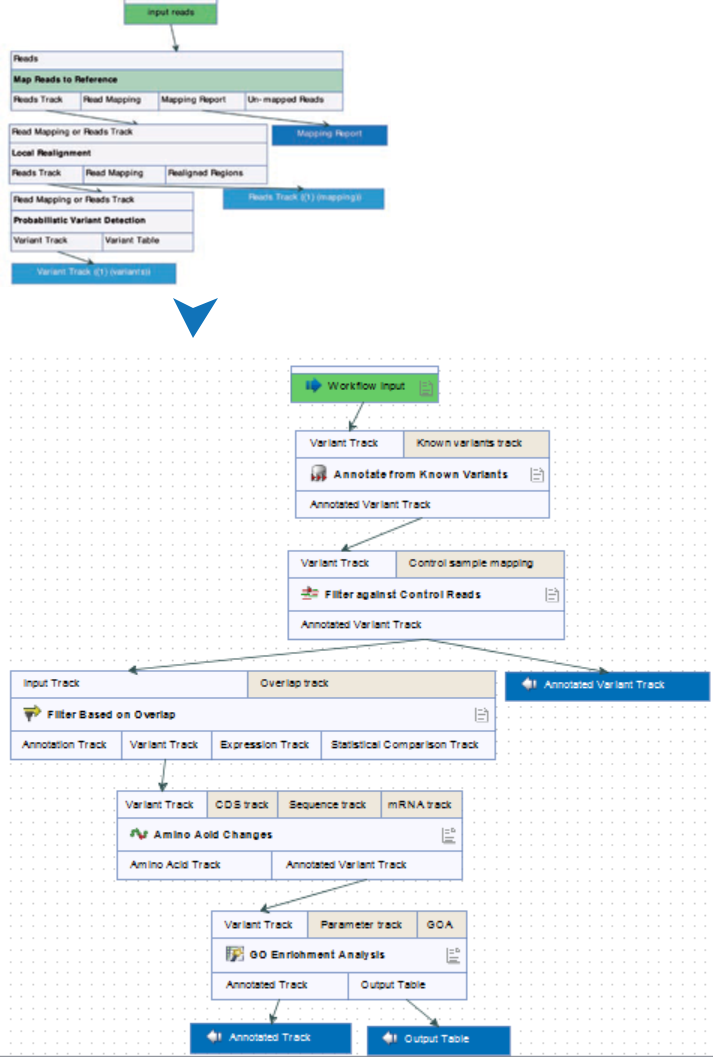
图1 数据分析的所有步骤能够组合成两个相继的分析流程,如上图所示。图中上半部分的分析流程将三种牛测序数据进行Read Mappings和变异识别,下半部分的分析流程是筛选和注释瘤牛特有的变异。


