

AI-enriched COMPUTATIONAL SIMULATION
AI计算模拟
以AI、大数据分析及数字化工作流为基础的综合计算模拟解决方案
VASP软件介绍
VASP(Vienna Ab-initio Simulation Package)是维也纳大学Hafner小组开发的进行电子结构计算和量子力学-分子动力学模拟软件包。它是目前材料模拟和计算物质科学研究中流行的商用软件之一。VASP是基于贋势平面波基组的第一性原理密度泛函计算程序,VASP软件作为目前国内国际上的第一性原理计算软件,可以研究多种体系,包括金属及其氧化物、半导体、晶体、掺杂体系、纳米材料、分子、团簇、表面体系和界面体系等。
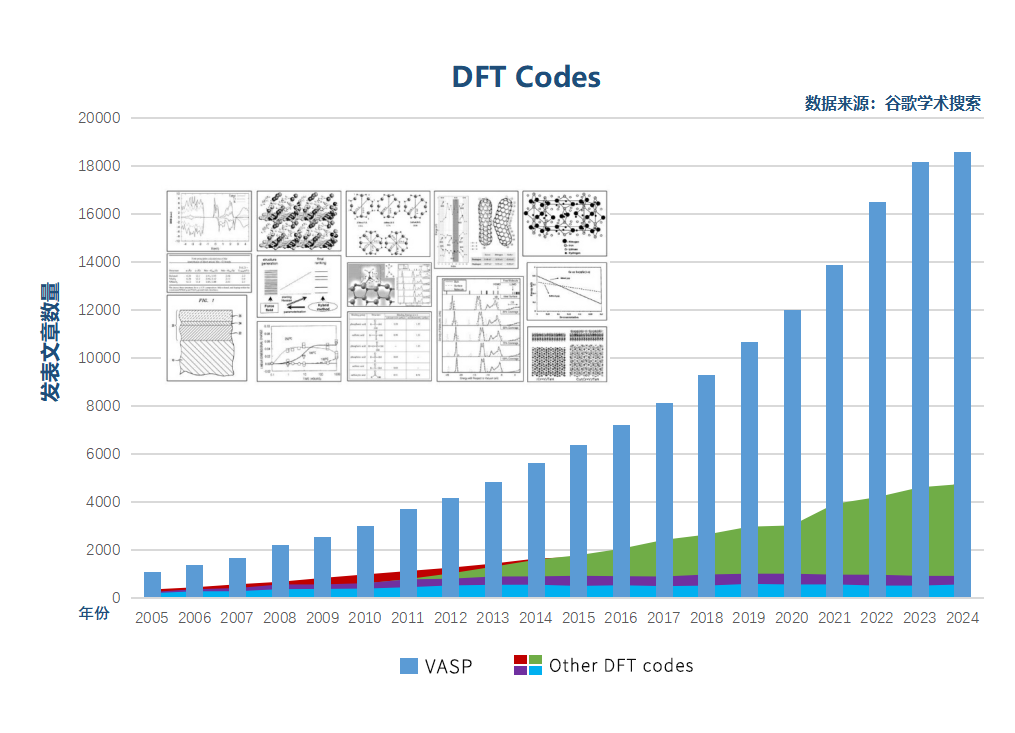
VASP不仅能够计算得到各种体系的平衡结构和能量,而且还能够对材料的电子性质进行准确的预测,深度剖析材料的各种理化性质。VASP软件功能强大,性能稳定,每年在许多国际核心期刊上发表上万篇文章(知名的杂志诸如Science、Journal of the American Society、Nano letters、Physical Review Letters等)。同时,VASP支持CPU和GPU计算,可以使用较小的内存实现大规模的高效率并行计算。
VASP能够计算的性质
VASP软件的基本原理是在密度泛函理论(DFT)框架内采用平面波基组展开的方法求解Kohn-Sham方程。VASP采用周期性边界条件(或超晶胞模型), 基于密度泛函和赝势理论对原子、分子、表面、团簇等多种体系进行几何结构优化得到稳定构型,进而获得各种结构参数和能量。同时,还能够计算多种电学、光学、磁学等性质。
Ø 几何性质 结构参数(键长、键角、晶格参数、原子位置),稳定构型等 | Ø 能量 体系总能量、表面能、界面结合能、吸附能、缺陷形成能 |
Ø 电子性质 电子态密度,能带结构,电荷密度分布,电子局域化函数(ELF),Bader Charge等 | Ø 光学性质:介电函数矩阵 Ø 计算材料的激发态(GW准粒子修正) Ø 计算X-ray吸收光谱(XAS) |
Ø 磁学性质 自旋极化、共线和非共线性磁性,自旋轨道耦合 Ø 机器学习(MLFF) | Ø 从头分子动力学模拟 Ø 计算材料的激发态(GW准粒子修正) Ø 电声耦合方法 |
VASP的赝势库
VASP软件是基于贋势和平面波基组的第一性原理密度泛函计算程序。VASP使用的是平面波基组,电子与原子核之间的相互作用使用投影缀加波贋势(Projector Augmented Wave,PAW)方法描述,从而进行量子力学计算。VASP采用PAW贋势,使得平面波基组尺寸非常小,描述原子一般不超过100个平面波基组,大多数情况下每原子50个平面波就能得到可靠结果。
VASP软件中的PAW贋势分为以下几种分类方法
按贋势支持的泛函种类分为GGA和LDA;
按原子中是否处理的半芯态(semi-core),分为A,A_sv,A_pv和A_d;
按赝势文件中截断能参数ENMAX的大小,分为普通赝势A,软赝势A_s(精度较低,速度较快)和硬赝势A_h(精度较高,速度较慢);
用于支持GW/RPA计算的贋势为A_GW。
VASP支持的泛函
Ø 局域密度近似(LDA)广义梯度近似(GGA) LDA、GGA-AM05、GGA-PBEsol、GGA-PBE、GGA-rPBE、GGA-BLYP | |
Ø 杂化泛函 HSE06、PBE0、B3LYP、DDH/DSH | |
Ø Meta-GGA泛函 revTPSS、TPSS、M06-L、MBJLDA、SCAN、rSCAN、r2SCAN、MS0、MS1、MS2、M06-L、MBJLDA | Ø 范德华密度泛函 optB86b-vdW,optB88-vdW,optPBE-vdW,rPW86-vdW2,revPBE-vdW、BEEF-vDW、SCAN+rVV10,rev-vdW-DF2 |
VASP的算法特征
在密度泛函理论(DFT)框架内求解Kohn-Sham方程
在Hartree-Fock(HF)的近似下求解Roothaan方程
支持格林函数方法(GW准粒子近似,Low Scaling GW,ACFDT-RPA)
微扰理论(二阶Møller-Plesset),支持屏蔽交换(Screened Exchange)。
支持从头分子动力学,AIMD
使用高效的矩阵对角化技术求解电子基态。在迭代求解过程中采用了Broyden和Pulay密度混合方案加速自洽循环的收敛
自动确定任意构型的对称性。利用对称性方便设定Monkhorst-Pack特殊点,可用于高效地计算材料和对称团簇。Brillouin区的积分使用模糊方法或Blöchl改进的四面体布点-积分方法,实现更快的k点收敛
支持scaLAPACK大尺度快速计算
支持自旋极化、自旋轨道耦合、非共线性磁性
支持对d或f轨道强关联作用计算的DFT+U方法
支持Space-time计算方法
基于Zacharias-Giustino方法考虑电子-声子耦合,计算X-ray吸收光谱(XAS);光谱发射率、总发射率关于温度的函数及黑体辐射关于频率的函数。
能够实现大规模的高效率并行计算,支持多核多节点并行计算,对核数和节点数均没有限制,支持单用户多用户同时使用。



