
近日,我们迎来了IPA发布的2023年新版,此版本新增了细胞类型的识别、自定义通路的因果预测、参考数据集的设置等新功能。目前,IPA知识库数据量已超过1230万条,收录的数据集已超过13万个。
一、功能更新
1. 根据网络和通路上的某组基因识别潜在的细胞类型
在这个新版中,IPA可以预测与用户的网络或通路上的基因相关的细胞类型。这一预测功能基于的是用户所查看通路上的某组基因与特定细胞类型中表达相对较高基因的富集。底层的细胞类型表达数据来自人类蛋白质图谱(https://www.proteinatlas.org)。
以来源于自然杀伤单细胞簇(来自人类胎儿肝脏,PMID 31597962)的表达数据为例,将Cells and Tissues(如图1中蓝色CT所示)叠加到网络中,可以清晰地看出该网络富含自然杀伤细胞基因(p值2.04E-20)(图1)。
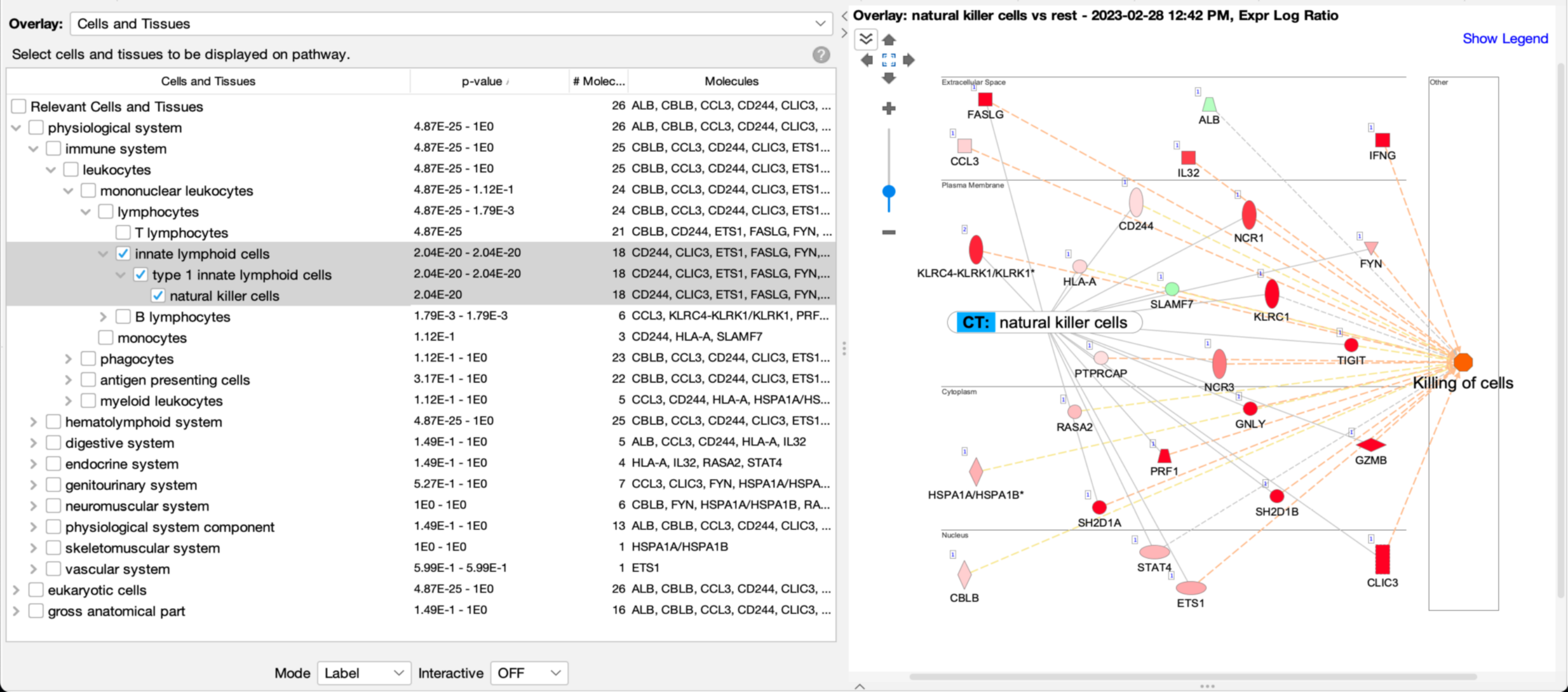
图 1:网络中自然杀伤基因的富集。
根据Ingenuity Ontology,细胞类型被分为三个主要分支:生理系统、真核细胞和大体解剖部分。某个细胞类型通常会出现在这些主要分支的两个或三个分支中。例如,在图1的例子中,在免疫系统下(即生理系统分支内)可以找到自然杀伤细胞,如图2所示,在真核细胞分支的血细胞下也可以找到自然杀伤细胞。

图 2:自然杀伤细胞也被归到Ingenuity Ontology的真核细胞分支。
在这个IPA版本中,细胞类型数据来自“RNA单细胞类型组织簇”数据,代表了人类蛋白质图谱中79个不同的细胞类型簇。开发团队计划在未来的版本中添加更多其他来源的细胞和组织类型。
2. 在核心分析中对My Pathways进行因果评分
借助这一新功能,用户可以在My Pathway上设置激活或抑制基因的模式,然后IPA可以通过将该模式与数据集中分子(指analysis-ready molecules)的差异表达进行比较,对其进行评分。
由此,IPA可以预测My Pathway在用户所分析数据集的背景下是被激活还是被抑制。每个节点的激活状态(红色或绿色)可以通过叠加分析或数据集来设置,可以手动使用MAP(分子活性预测)功能中的红色或绿色油漆桶,也可以将油漆桶与叠加的分析或数据集一起进行组合设置。
图3举例了在IPA中创建的一个My Pathway,它描绘了几个关键的上皮-间充质转化相关基因和生物功能,基因节点已经用MAP油漆桶进行了着色。保存该通路并将其批准用于评分,就可以在未来的核心分析中对该通路进行评分。
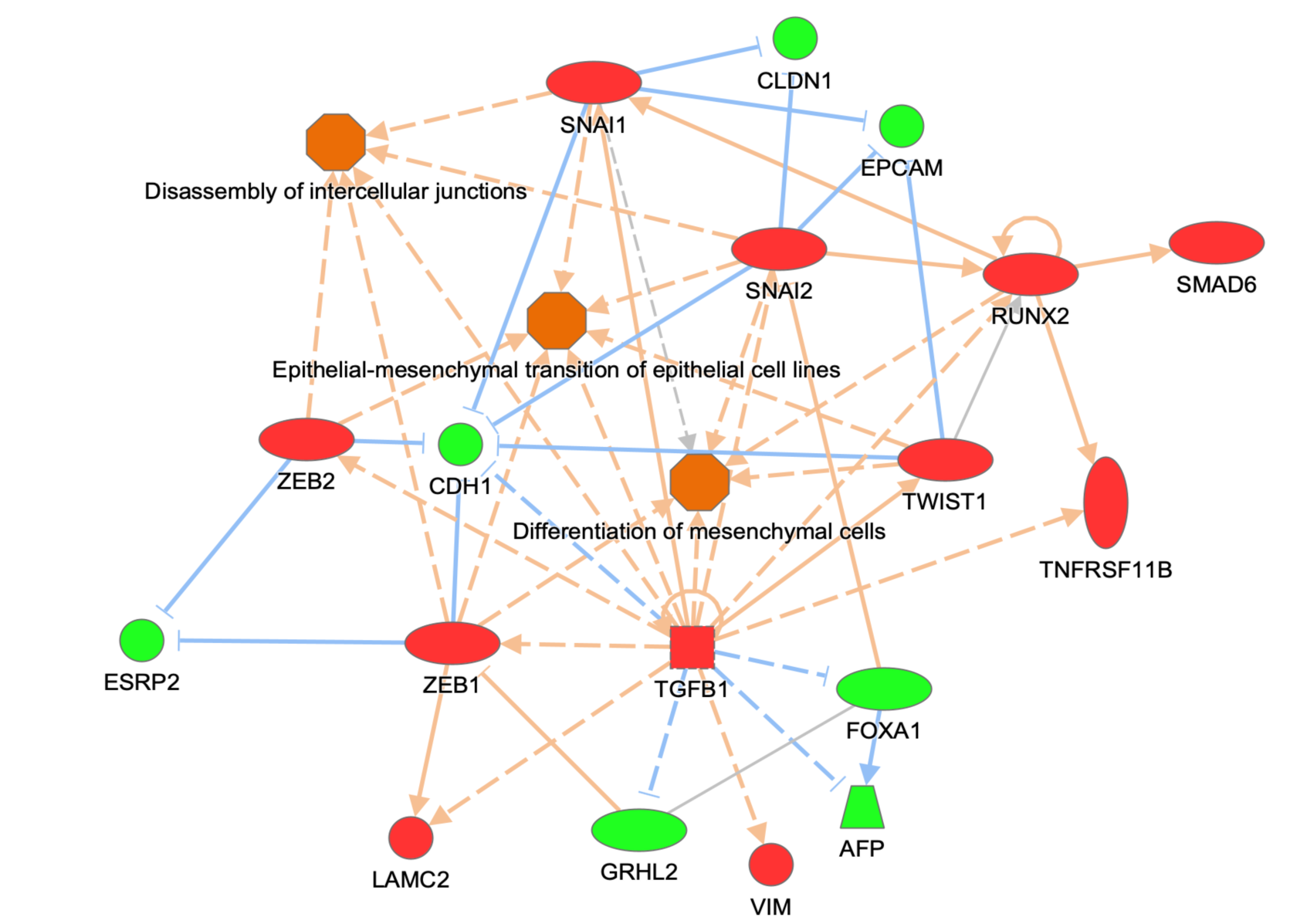
图 3:自定义的My Pathway,其中的节点可由用户指定为激活(红色)或抑制(绿色)。
评分是使用z-score算法完成的,类似于经典通路的评分方式,即将数据集中分子的上调或下调状态与每个My Pathway中匹配分子的活性状态进行比较。图4显示了My Pathways选项卡,展示了对claudin-low型乳腺癌细胞系与管腔细胞系(PMID 20813035)表达数据进行的核心分析。
经过IPA预测,在侵袭性癌症细胞系中,自定义的EMT(上皮-间质转化)通路被激活,这也正是这些细胞预期的结果。z-score计算所得为正值,因为数据集中的实际表达方向(如图4表中的第四列Measurement所示)与所保存的My Pathway中指定的预期方向(如图4表中的第七列Expected所示)相匹配。
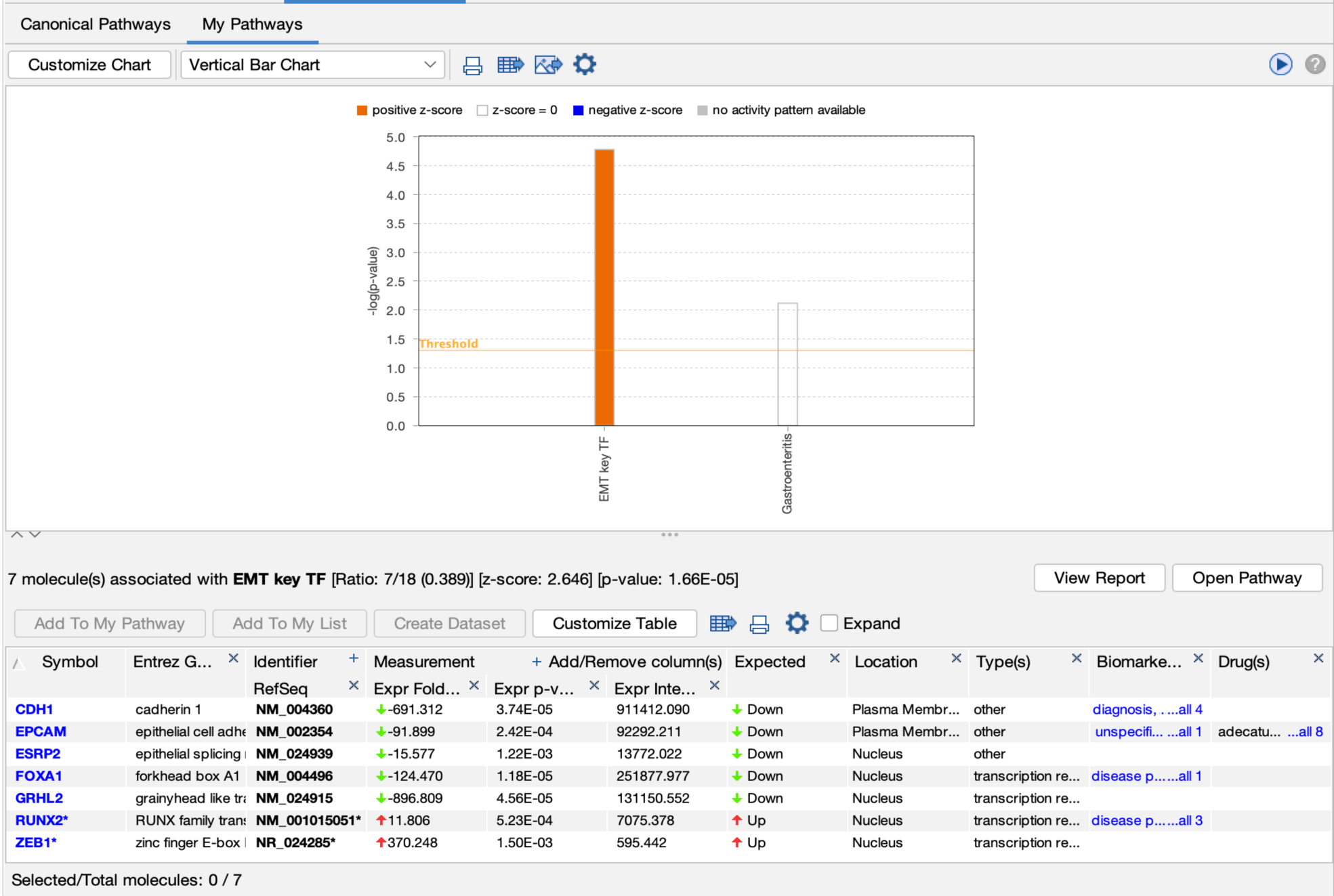
图 4:对名为“EMT key TF”的自定义My Pathway进行因果评分。橙色柱子表明,在侵袭性乳腺癌症细胞系的表达分析中,该通路被预测为激活(该通路的z-score为2.646)。
用户可以创建任意想要的通路,并了解它在实验环境中是如何受到影响的。而且,通路上的基因不需要具备关系连接,用户也可以通过修改经典通路或其他的IPA通路,生成自定义的通路。
3. 将“用户数据集”设置为参考集
在分析数据集时,用于统计计算的理想参考基因集应该是与实验环境中测量(或可以测量)的基因集接近的基因集。假设您正在分析一个包含400个基因的数据集,那么参考集应该是这400个基因,或者是手头的实验条件下可测量基因集的子集。如果在实际的实验中只能测量这400个基因的变化,那么将参考集设置为基因组中所有的基因在统计上就是不正确的。
举个例子,如果您正在进行小鼠肾组织中的全转录组数据分析,那么理想情况下,参考集应该是您能够可靠测量的实验中所有基因的集合。例如在至少一个样本中RPKM值超过某个阈值(RPKM>1)的基因,这样以来,参考集就应该被设定为“小鼠肾脏表达的基因”,而不是基因组中所有的基因,因为其中一些基因在小鼠肾脏中根本不表达。
用户可以上传未筛选的整个可检测到的分子集到IPA中,然后在分析数据时,将“用户数据集”设置为参考集。然而,在老版本的IPA中,用户在创建分析时往往很容易忘记进行这个设置,顺手就将整个基因组用作参考集。
在这个新版IPA中,用户可以在数据集上传期间,将参考集设置为“用户数据集”,因为此时用户更可能记得去正确地进行参考集的设置。这个新功能可以避免用户在分析中不小心使用不太理想的参考集。
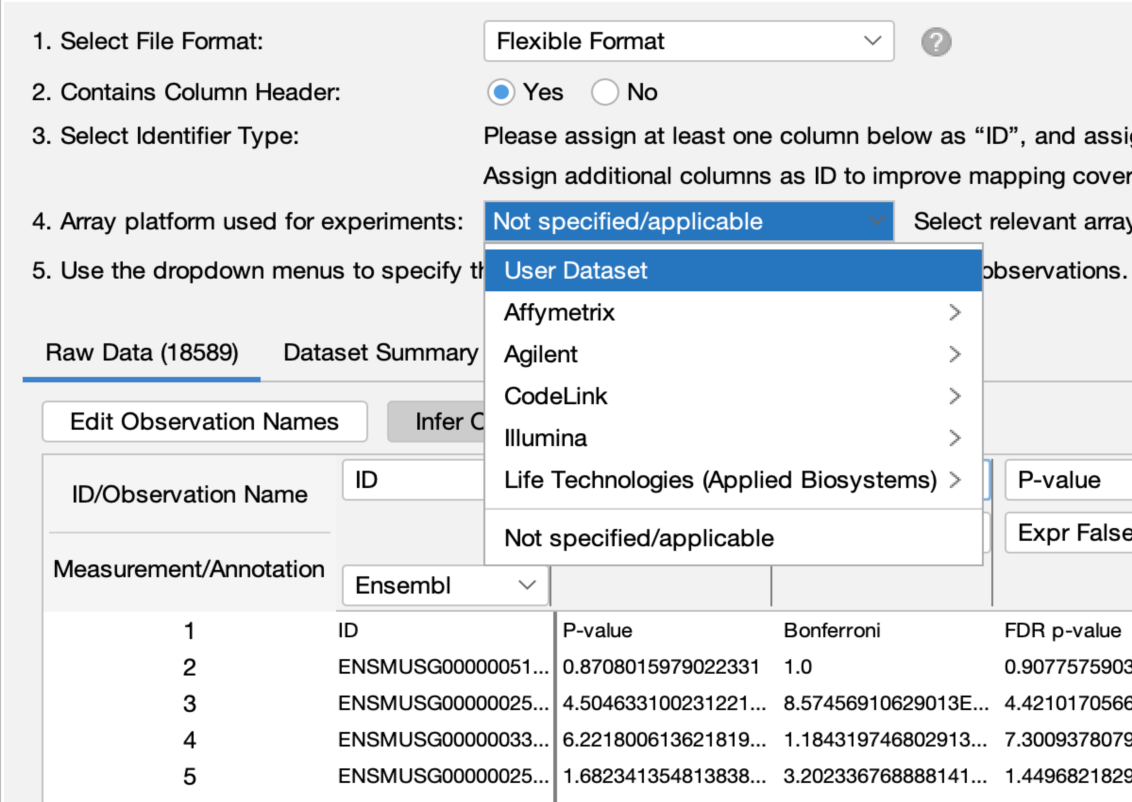
图 5:在数据集上传时将参考集设置为“用户数据集”(即图中选中的User Dataset)。
请注意,如果您的数据集仅代表实验中显著差异表达的基因,则不应使用“用户数据集”作为参考集。在这种情况下,如果您在分析时没有设置更严格的cutoff值,那么统计数据就是不正确的,因为此时待分析的基因(analysis-ready genes)和参考集之间没有区别。这些统计计算的目的是呈现参考集中一小部分基因的富集情况。
4. 图形摘要的图例
查看图形摘要Graphical Summary(核心分析中的一个选项卡)时,界面的右上角会出现一个该选项卡专用的图例,如图6所示。
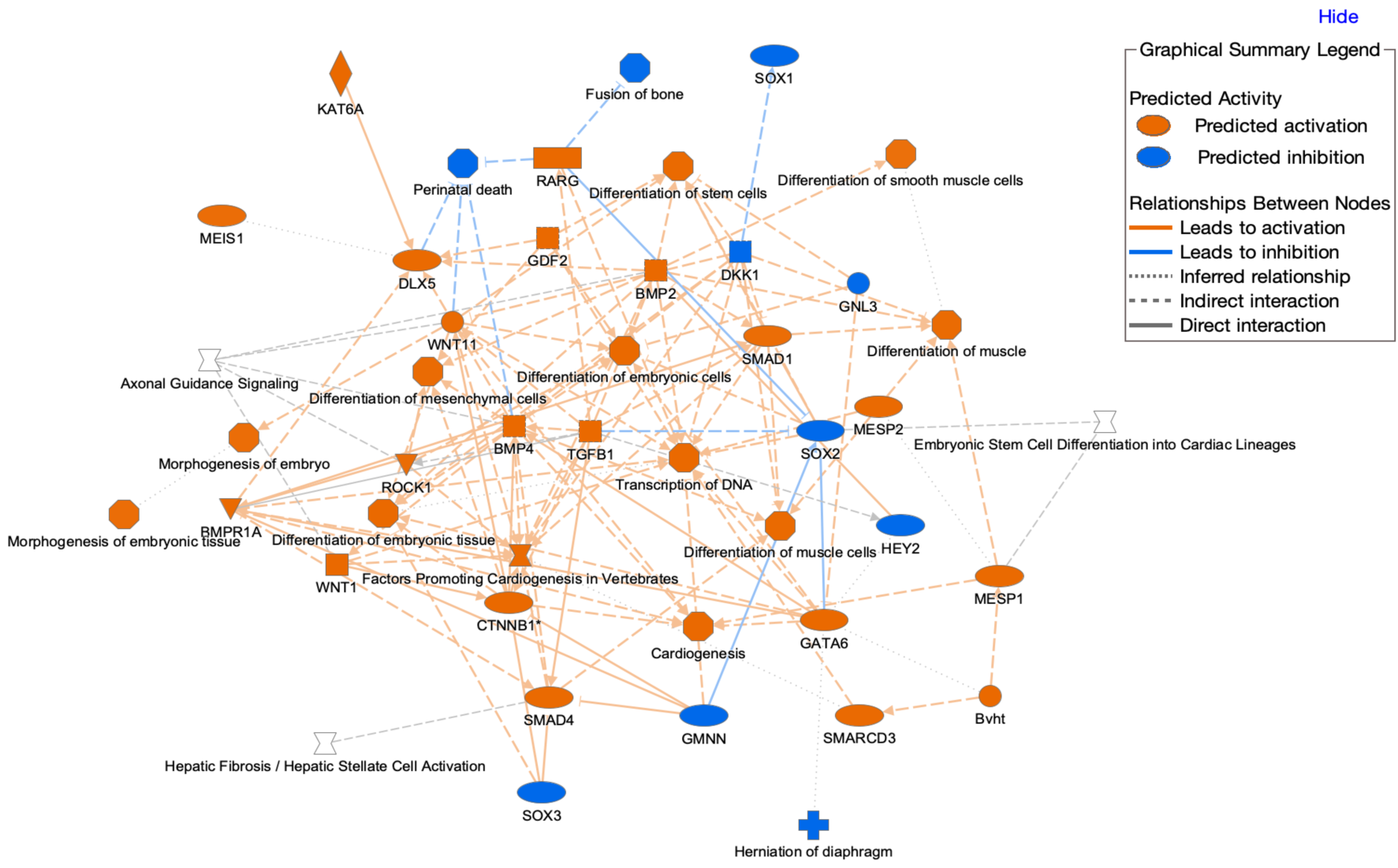
图 6:图形摘要的图例(可下载高分辨率图片文件用于文献发表)。
二、内容更新
本季度新增了超过26万项新发现,使得IPA知识库数据量超过1230万条。新增的内容包括BioGrid的蛋白-蛋白相互作用、IntAct的蛋白-蛋白相互作用、ClinVar的癌症突变发现、ClinicalTrials.gov的靶点-疾病发现和药物-疾病发现、Gene Ontology的发现、在线人类孟德尔遗传(OMIM)的基因与疾病关联、COSMIC的癌症突变发现、人类蛋白质图谱(THPA)的基因-细胞类型发现等。
在Analysis Match、Activity Plot、Pattern Search、Land Explorer等功能中,用户可以探索超过13万个数据集(具体数据统计如下表),或是将它们与自己的数据集进行比较分析。

图 7:Land数据集统计表。
IPA的内容团队和开发团队一直以来都在不断提升和完善知识库和功能,源资科技一直以来都在不断为广大的用户带来优质的解决方案和支持服务。欢迎联系我们体验IPA的新功能!


