
1 案例背景
对于电动车辆应用,锂离子电池(LIBs)能量密度的重要性不言而喻。当前商用LIB阴极材料如层状锂过渡金属氧化物(LiMO2,M = Ni, Mn, Co)由于其较低的容量,成为限制因素。为了克服这一问题,研究人员开始关注富锂层状氧化物(LLOs),即Li1+xM1-xO2 (M = Mn, Ni, Co) 的复合材料。这些材料因额外的阴离子氧化还原活性而表现出超过250 mAh g−1的高放电容量。然而,LLOs也面临一些挑战,包括首次循环中较高的不可逆容量损失、较差的倍率性能、长期循环中的容量衰减和平均放电电压下降、金属阳离子迁移以及气体释放等问题。这些问题主要与LLOs中的Li2MnO3组分有关,该组分在充电过程中容易发生氧释放及结构不稳定。为了改善这些问题,一般采用了掺杂策略来抑制结构变化,从而提高容量保持率并缓解电压衰减。此外,理论计算作为一种强有力的工具,可以系统地研究掺杂对材料基本性质的影响,以识别合适的掺杂元素。本案例中, 大众公司考虑到Li2MnO3在LLOs中的重要作用,尤其是它所涉及的阴离子氧化还原活性、结构变化和氧释放等问题,本案例旨在深入探讨特定阳离子掺杂如何影响脱锂过程、氧化还原电位、电子结构以及氧阴离子的氧化程度。
2 建模与计算方法
作者通过InfoMaticA中搜索得到Li2MnO3(空间群为C2/m)的结构,并采用一个M原子(M=Ti,Al)替换一个Li或一个Mn原子。考虑到Li2MnO3结构中锂有多种不同位点,如图1所示,M替换锂原子以及锂离子脱出嵌入时,会有多种不同的情况,导致计算模型数量也极多。因此为了得到最稳定的替换位点模型,以及锂离子的脱出嵌入模型,作者采用MedeA UNCLE基于少量MedeA VASP计算,对最稳定结果进行搜索预测,降低整体工作量,提高搜索效率。在MedeA VASP计算中,截断能采用520 eV,K点间隔为0.4 Å-1。体系能量与原子受力收敛标准分别为10-5 eV,0.02 eV/Å。对于过渡金属元素,采用U+J方法处理3d轨道电子,Mn与Ti的U+J取值分别为,4.3+0.8 eV,4.0+0.8 eV。
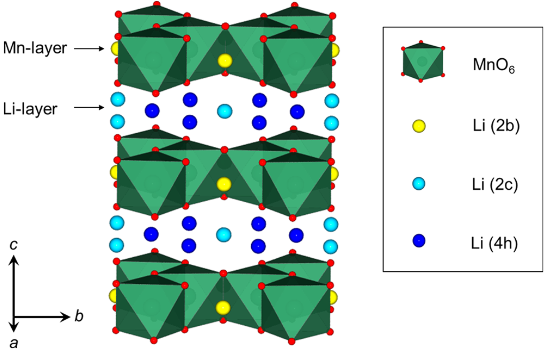
图1 Li2MnO3晶体中不同锂的位置以及MnO6多面体。
3 结果与讨论
3.1 脱锂基态相图
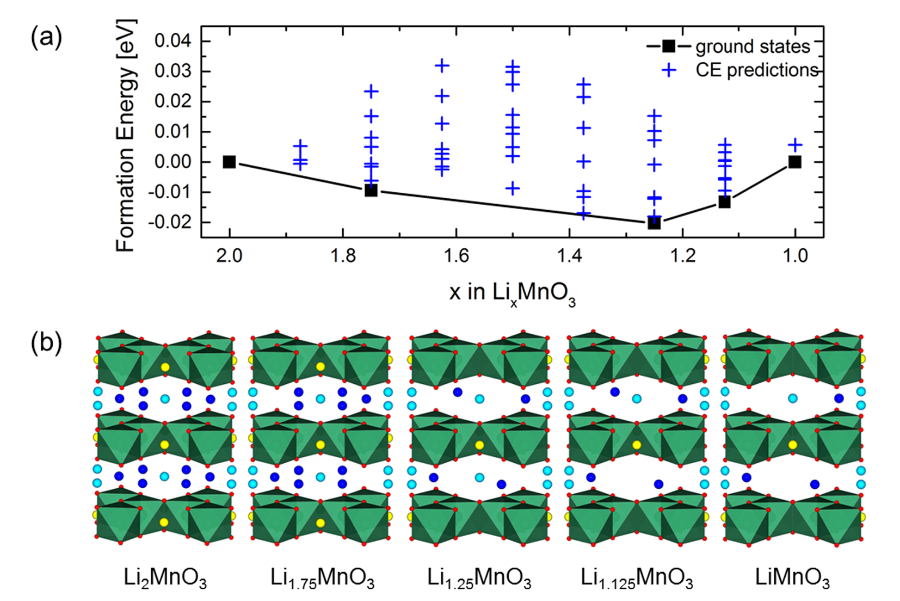
图2 MedeA UNCLE搜索预测Li2MnO3脱出锂过程。(a)脱锂结构的形成能,黑色曲线表示基态,(b)不同浓度下最稳定结构
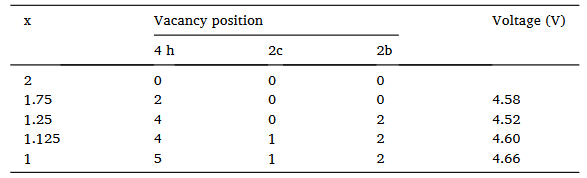
作者基于MedeA VASP采用MedeA UNCLE计算并搜索预测了Li2MnO3脱锂过程的基态相图,如图2所示。从图中可以看到五个基态最稳定的结构,对应LixMnO3中x分别为2,1.75,1.25,1.125,1。当x取1.25时,形成能最低为-20.2 meV。脱出锂时,对应的电压如表 1所示。整个脱锂过程,电压范围为4.58~4.66 V,与目前实验电压平台值4.5 V一致。
表 1 Li2MnO3脱出锂时最稳定结构的电压
作者采用MedeA VASP计算不同元素取代Li或Mn时相对于纯Li2MnO3的形成能。可以看到,当Al或Ti掺杂替换后,基态结构的相对形成能都低于0,说明Al或Ti掺杂具有稳定Li2MnO3的效果,并且Al或Ti取代Mn后的相对形成能更低,因此Al或Ti更倾向替换Mn而非Li。同时作者计算了Al或Ti取代其它位点的Li的形成能,发现Al取代2b位置的锂,Ti取代4h位置的锂时,最稳定。
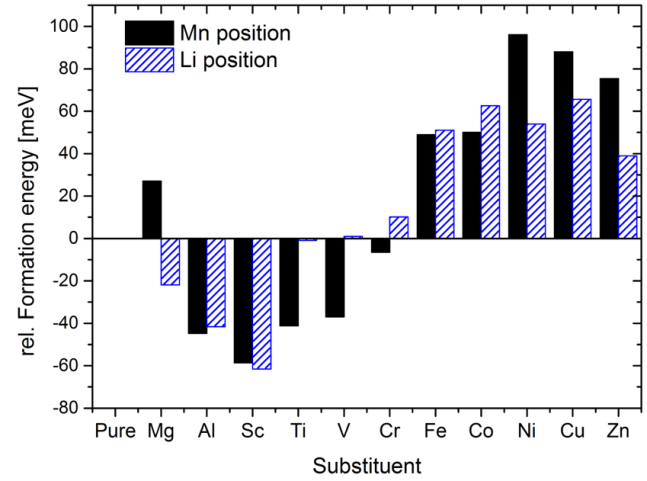
图3 不同元素取代Li或Mn时相对于纯Li2MnO3的形成能,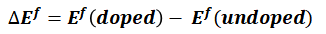
在一些已有文献中,为了简化计算,掺杂体系的脱锂顺序一般与纯体系的掺杂相同,因而忽略了掺杂原子的作用,得到的脱锂结构并非热力学上最稳定的结构,因此相应的结论也是不准确。为此,作者基于MedeA VASP采用MedeA UNCLE计算并搜索预测在Al、Ti掺杂情况下锂脱出的基态相图,如图 4所示。
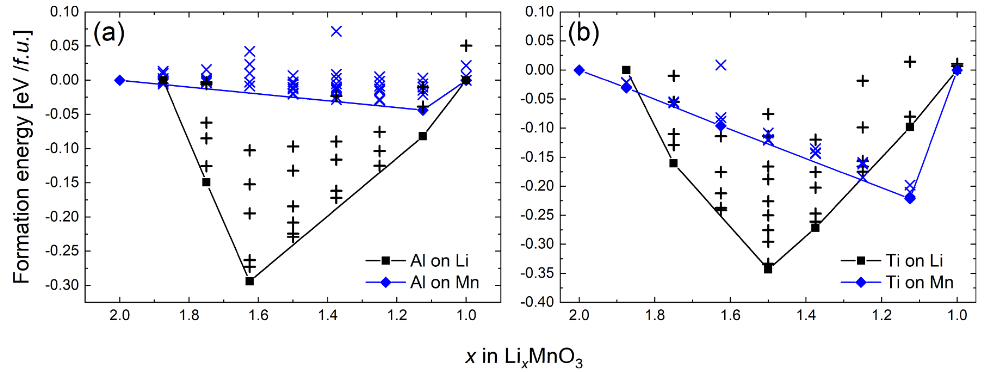
图 4 MedeA UNCLE搜索预测Al或Ti掺杂时,Li2MnO3脱锂后结构的形成能。(a)Al掺杂,(b)Ti掺杂
相比纯Li2MnO3体系脱锂的形成能(-20~0 meV)而言,掺杂后脱锂结构的形成能可达-350 meV。并且,掺杂替换的元素(Li,Mn)不同时,脱锂结构的形成能相差极大。替换Li之后,脱锂结构的形成能远低于替换Mn的情况。
3.2 电化学性质与电子结构
基于上一部分最稳定的脱锂结构,作者采用MedeA VASP进一步分析其电化学性质与电子结构。图 5是不同掺杂体系的还原电势。从图 5(a)可以发现,Ti掺杂初始电势最低;当掺杂原子替换Mn时,对电势影响很小。在整个脱锂浓度范围内,所有电势都接近4.5 V;当掺杂原子替换Li时,脱锂浓度初期,电势较低。为了解释初始电势低的问题,作者计算了体系的分波态密度。

图 5 (a)不同掺杂体系的还原电势,(b)(a)图中4.2~4.8 V范围内曲线图
图 6为无掺杂体系,替换Mn/Li体系的分波态密度。从图中可以看到,当替换Mn时,体系的分波态密度与无掺杂体系基本相同,说明掺杂替换Mn对Li2MnO3体系的电子结构影响极小。相反,对于掺杂替换Li来说,电子结构变化极大:(1)费米能附近出现亲的极性态密度,由Mn 3d与氧的2p轨道形成;(2)具有共价性的元素替换锂之后,导致Mn4+被还原成Mn3+,氧化开始时Mn3+最先被氧化,导致体系初期的还原电势低;(3) MnO6多面体由于Jahn-Teller效应产生畸变,Mn-O 键长从1.93 增加到1.95,2.21 Å。作者进一步计算了,掺杂对脱锂过程中氧空位形成能影响,如图 7所示。随着脱锂过程进行,锂含量逐渐降低,纯Li2MnO3中氧空位形成能从正变负,逐渐降低,表明氧空位形成更加容易。进行Al/Ti掺杂后,随锂含量降低,锂空位形成能也逐渐降低,但总体来说高于纯Li2MnO3体系,说明Al/Ti掺杂能抑制氧空位形成。
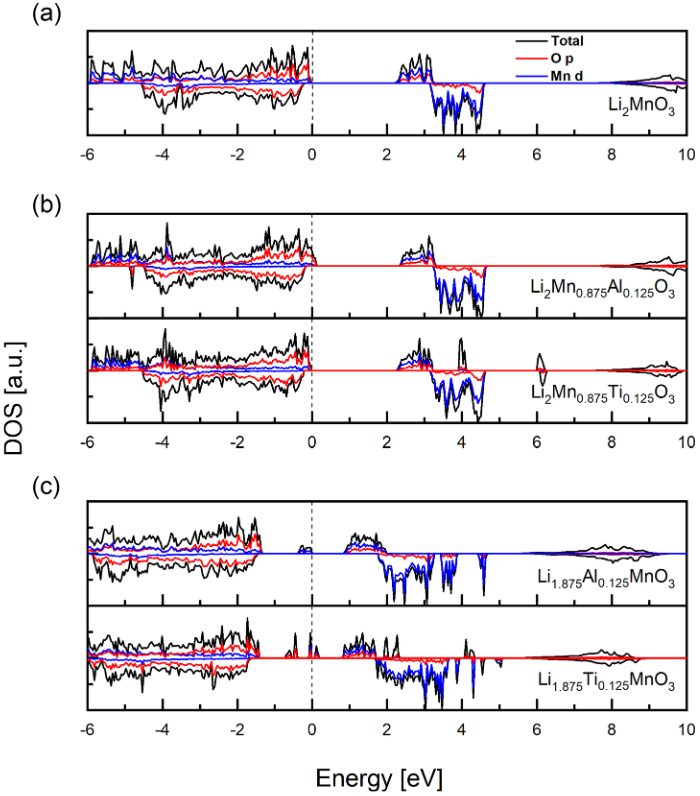
图 6 (a)无掺杂体系,(b)替换Mn体系,(c)替换Li体系的分波态密度
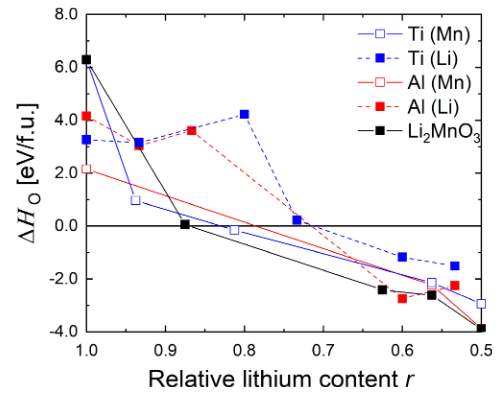
图 7 不同体系中氧空位的形成能与锂含量之间的关系
4 总结与展望
本案例研究了阳离子Al/Ti掺杂对富锂层状氧化物(LLOs)的富锂锰氧化物(Li2MnO3)结构的氧化还原电位、电子结构和部分阴离子氧化的影响。为了获得热力学上有意义的结果,采用集团展开法计算了纯Li2MnO3以及掺杂替换Li/Mn后的脱锂结构。掺杂后脱锂结构形成能显著降低说明,掺杂剂强烈影响锂的脱出嵌入顺序,表明了在锂离子脱嵌研究中考虑集团展开法的重要性。掺杂替换Li时,对初始氧化还原电位和电子结构影响较大,这是由于用较高价态的阳离子替代锂导致Mn⁴⁺离子被还原从而降低了氧化还原电位。另外,Al/Ti掺杂显著减少了脱锂过程中氧空位的形成能,能有效抑制氧气释放的趋势。这项理论研究表明,掺杂可能是提高这类前景极高材料的稳定性的方式之一。在计算掺杂结构的脱锂过程时,生成热力学稳定的脱锂结构是十分重要的,并为LLOs的电化学性质提供电子结构层次的理解。
参考文献:
Jens Matthies Wrogemann, Tanja Graf, Jonathan E. Mueller , Thomas D. Schladt. Theoretical investigations of selective cation doping as a novel design strategy for high-capacity lithium-rich cathode materials [J]. Computational Materials Science, 2022, 204:11177.
使用MedeA模块:
MedeA Environment
MedeA VASP
MedeA UNCLE


